
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of the role of hydrogen bonding in ionic liquid-like salts with both N- and S-soft donors†
Olivier
Renier
 a,
Guillaume
Bousrez
a,
Volodymyr
Smetana
a,
Anja-Verena
Mudring
*ab and
Robin D.
Rogers
*ac
a,
Guillaume
Bousrez
a,
Volodymyr
Smetana
a,
Anja-Verena
Mudring
*ab and
Robin D.
Rogers
*ac
aDepartment of Materials and Environmental Chemistry, Stockholm University, 10691 Stockholm, Sweden. E-mail: mudring@iastate.edu
bDepartment of Chemistry and iNANO, 253 Aarhus University, 8000 Aarhus C, Denmark
cDepartment of Chemistry & Biochemistry, The University of Alabama, Tuscaloosa, AL 35487, USA. E-mail: rdrogers@ua.edu
First published on 5th December 2022
Abstract
In search of ionic liquids (ILs) with multiple types of soft donor atoms capable of preferentially complexing a range of soft metal ions over harder ions, we investigated structural clues to the role of hydrogen bonding in IL behavior through a series of salts with anions containing both N- and S-donor atoms based on azole thiolates. Reaction of equimolar amounts of triethylamine (Et3N) or diisobutylamine (DBA) with 1-phenyl-1H-tetrazole-5-thiol (PhTzSH), 1-methyl-1H-tetrazole-5-thiol (MeTzSH), or 5-methyl-1,3,4-dithiazole-2-thiol (MeDiTSH) yielded [Et3NH][MeTzS] (1), a yellow liquid, and the low melting yellow solids [DBAH][MeTzS] (2), [Et3NH][PhTzS] (3), [DBAH][PhTzS] (4), [Et3NH][MeDiTS] (5), and [DBAH][MeDiTS] (6). Thermal analysis revealed that all of them qualify as ILs with melting points below 100 °C. Single crystal X-ray structure analysis of 2–6 revealed the presence of an extensive H-bonding network that includes the rare N–H⋯S hydrogen bonds in 3, 4, and 6. These weaker interactions appear to significantly influence thermal behavior, where strong bonding leads to higher melting temperatures and lower decomposition points.
1. Introduction
Separations involving f-elements are currently of considerable interest in a variety of fields including nuclear chemistry,1,2 environmental remediation,3 and recovery of lanthanides from electronic waste.4,5 The separation of f-elements is extremely difficult and finding an easy and cheap method to achieve this is becoming ever more important as they are key constituents in materials for energy efficient applications such as in magnets for wind turbines and e-vehicles as well as consumer-related.6–8 This is particularly true for separations of heavier 5f-elements from 4f-elements in nuclear waste remediation, where although both hard donor complexants will sometimes work; the softer donor complexants have been used to separate transuranic elements from their lanthanide analogs.2 Controlling the number and type of soft donor atoms in a given complexant may offer the possibility to selectively extract 4f- from 5f-metals or vice versa.9–14We and others have explored a range of new f-element separations using ionic liquids (ILs) as both solvents and complexants.4,15,16 ILs can easily accommodate many desired functionalities separately on their cationic and anionic parts or jointly on a given ion while still allowing additional chemical tuning towards the required physical properties of a given separation (melting point, density, viscosity, etc.).17 While there have been many studies to tune IL physical properties for separation, there have been comparatively few which have focused on tuning the hard/soft donor balance needed to execute separations of lanthanides and actinides, which have similar hardness.18
To begin to build a library of soft donor salts and learn more about features of mixed soft donor anions that would be amenable to forming low melting salts, we began this study of thiolate anions formed by deprotonation of 1-phenyl-1H-tetrazole-5-thiol (PhTzSH), 1-methyl-1H-tetrazole-5-thiol (MeTzSH), and 5-methyl-1,3,4-dithiazole-2-thiol (MeDiTSH).
In general, thiolate ILs have rarely been investigated and little is known about their intra- and intermolecular interactions. Azole thiols present the extra advantage of containing two soft donor atoms, sulfur and nitrogen,19 which can further be exploited towards metal-specific interactions.20 The complexation of Hg(II) via the sulfur atom by some derivatives such as the 5-mercapto-1-methyltetrazole has been reported,21,22 while the presence of nitrogen as a second soft donor could promote the complexation of other metal classes such as f-elements. Thiols in hydrophobic ILs have also been recommended as potential ligands to extract toxic heavy metals.23–26
We hypothesized that these types of ILs could be better understood and designed following the approach of anti-crystal engineering, i.e., by the deliberate avoidance of any structural features that favor interaction between cation and anion and thus intricate crystallization.27,28 For this purpose, a series of salts with two small amines, triethylamine (Et3N) and diisobutylamine (DBA), was prepared and structurally characterized. Binary and ternary amine cations have been involved to evaluate the H-bonding factor and compare their ability to yield crystalline salts. Here, we report our initial results obtained by simple acid/base reactions of MeTzSH, PhTzSH, and MeDiTSH with Et3N and DBA.
2. Experimental section
2.1 Chemicals and materials
Triethylamine (Et3N, ≥99.5%), diisobutylamine (DBA, 99%), 5-mercapto-1-methyltetrazole (MeTzSH 98%), 1-phenyl-1H-tetrazole-5-thiol (PhTzSH, 98%), 5-methyl-1,3,4-dithiazole-2-thiol (MeDiTSH, 99%) and methanol (99.8%) were all purchased from Sigma-Aldrich (Steinheim, Germany) or Alfa Aesar (Karlsruhe, Germany). Et3N and DBA were distilled over KOH before use.2.2 Synthesis
The respective amine (10 mmol, 1.0 eq.) and MeTzSH, PhTzSH, or MeDiTSH (10 mmol, 1.0 eq.) were added together with 20 mL of CH3OH (20 mL) in a 100 mL round bottom flask. The mixture was heated under reflux for 4 h and cooled to room temperature. The solvent was removed at 40 °C under reduced pressure using a rotary evaporator to yield the final product in quantitative yield. Single crystals suitable for single crystal X-ray diffraction (SCXRD) analyses were obtained by isothermal evaporation of dilute methanolic (2–4) or ethyl acetate (5–6) solutions.[Et3NH][MeTzS] (1): Yellow oil. 1H NMR (400 MHz, DMSO-d6): 1.19 (t, JH–H = 7.2 Hz, 9H), 3.12 (q, JH–H = 14.8 Hz, JH–H = 7.2 Hz, 6H), 3.66 (s, 3H). 13C NMR (100 MHz, DMSO-d6): 8.6, 32.6, 45.8, 167.1. νmax (cm−1): 2981, 2945, 2885, 2463, 1451, 1427, 1353, 1275, 1260, 1212, 1160, 1082, 1034, 967, 898, 836, 808, 792, 705, 553, 536, 519, 498, 466, 418. ESI TOF m/z (positive mode) 102.1041 (calculated m/z = 102.1283). ESI TOF m/z (negative mode) 114.9359 (calculated m/z = 115.0078). Tglass = −57 °C; Tdecomp = 164 °C.
[DBAH][MeTzS] (2): Yellow solid. 1H NMR (400 MHz, DMSO-d6): 0.90 (d, JH–H = 6.4 Hz, 12H), 2.02 (hept, JH–H = 20.4 Hz, JH–H = 13.6 Hz, JH–H = 6.8 Hz, 2H), 2.84 (d, JH–H = 7.2 Hz, 4H), 3.66 (s, 3H), 8.60 (bs, 2H). 13C NMR (100 MHz, DMSO-d6): 20.1, 25.1, 32.6, 54.6, 167.3. νmax (cm−1): 3004, 2964, 2872, 2788, 2557, 2452, 1626, 1464, 1425, 1404, 1392, 1357, 1271, 1167, 1105, 1020, 973, 877, 839, 805, 708, 518, 460, 425. ESI TOF m/z (positive mode) 130.1221 (calculated m/z = 130.1596). ESI TOF m/z (negative mode) 114.9359 (calculated m/z = 115.0078). Tmelt = 95 °C; Tcryst = 38 °C; Tdecomp = 118 °C.
[Et3NH][PhTzS] (3): Yellow solid. 1H NMR (400 MHz, DMSO-d6): 1.17 (t, JH–H = 7.2 Hz, 9H), 3.12 (q, JH–H = 14.4 Hz, JH–H = 7.2 Hz, 6H), 7.38 (t, JH–H = 7.2 Hz, 1H), 7.50 (t, JH–H = 8.0 Hz, 2H), 8.02 (d, JH–H = 8.0 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): 8.5, 45.8, 123.5, 127.4, 128.5, 136.6, 167.0. νmax (cm−1): 2985, 2944, 2631, 2474, 1593, 1493, 1466, 1434, 1412, 1376, 1350, 1309, 1287, 1261, 1213, 1162, 1113, 1081, 1062, 1037, 1010, 987, 921, 896, 836, 809, 769, 737, 720, 693, 684, 613, 567, 535, 509, 455, 413. ESI TOF m/z (positive mode) 102.1041 (calculated m/z = 102.1283). ESI TOF m/z (negative mode) 176.9084 (calculated m/z = 177.0235). Tmelt = 79 °C; Tglass = −40 °C; Tdecomp = 135 °C.
[DBAH][PhTzS] (4): Yellow solid. 1H NMR (400 MHz, DMSO-d6): 0.92 (d, JH–H = 6.4 Hz, 12H), 2.01 (hept, JH–H = 20.4 Hz, JH–H = 13.6 Hz, JH–H = 6.8 Hz, 2H), 2.81 (d, JH–H = 7.2 Hz, 4H), 7.37 (t, JH–H = 7.2 Hz, 1H), 7.49 (t, JH–H = 8.0 Hz, 2H), 8.03 (d, JH–H = 7.6 Hz, 2H), 8.43 (bs, 2H). 13C NMR (100 MHz, DMSO-d6): 20.0, 25.1, 54.5, 123.4, 127.3, 128.4, 136.7, 167.1. νmax (cm−1): 2959, 2875, 2759, 2541, 2438, 1596, 1498, 1466, 1408, 1373, 1352, 1329, 1309, 1285, 1208, 1173, 1159, 1079, 1065, 1042, 1015, 964, 934, 909, 877, 838, 807, 760, 717, 693, 682, 563, 519, 459, 426. ESI TOF m/z (positive mode) 130.1221 (calculated m/z = 130.1596). ESI TOF m/z (negative mode) 176.9084 (calculated m/z = 177.0235). Tmelt = 98 °C; Tglass = −15 °C; Tdecomp = 134 °C.
[Et3NH][MeDiTS] (5): Yellow solid. 1H NMR (400 MHz, DMSO-d6): 1.18 (t, JH–H = 7.2 Hz, 9H), 2.42 (s, 3H), 3.10 (q, JH–H = 14.4 Hz, JH–H = 7.2 Hz, 6H). 13C NMR (100 MHz, DMSO-d6): 8.7, 15.8, 45.7, 159.4, 187.4. νmax (cm−1): 3451, 3038, 2976, 2855, 2708, 2647, 1702, 1553, 1463, 1447, 1377, 1308, 1266, 1196, 1173, 1129, 1050, 1025, 970, 836, 761, 737, 656, 621, 584, 537, 430. ESI TOF m/z (positive mode) 102.1041 (calculated m/z = 102.1283). ESI TOF m/z (negative mode) 131.1935 (calculated m/z = 130.9738). Tmelt = 34 °C; Tglass = −41 °C; Tdecomp = 164 °C.
[DBAH][MeDiTS] (6): Yellow solid. 1H NMR (400 MHz, DMSO-d6): 0.92 (d, JH–H = 6.4 Hz, 12H), 1.94–2.00 (m, 2H), 2.39 (s, 3H), 2.77–2.80 (m, 4H). 13C NMR (100 MHz, DMSO-d6): 15.6, 20.2, 25.3, 54.6, 158.9, 185.9. νmax (cm−1): 3041, 2957, 2869, 2803, 2753, 2711, 2654, 2525, 2396, 2334, 1554, 1464, 1449, 1402, 1392, 1378, 1329, 1314, 1270, 1192, 1174, 1132, 1075, 1065, 1047, 1030, 1007, 965, 927, 857, 804, 756, 742, 653, 623, 611, 586, 538, 520, 462, 432. ESI TOF m/z (positive mode) 130.1221 (calculated m/z = 130.1596). ESI TOF m/z (negative mode) 131.1935 (calculated m/z = 130.9738). Tmelt = 95 °C; Tcryst = 90 °C; Tdecomp = 169 °C.
2.3 Instrumentation
1H and 13C nuclear magnetic resonance (NMR) spectra were recorded at room temperature in DMSO on a Bruker 400 MHz spectrometer equipped with a BBO probe (Bruker, Ettlingen, Germany). Chemical shifts are reported in delta (δ) units, expressed in parts per million (ppm). The following abbreviations were used for the observed multiplicities: s (singlet), d (doublet), t (triplet), q (quartet), hept (heptuplet), br (broad), m (multiplet for unresolved lines). 1H NMR chemical shifts were referenced to the residual solvent signal for DMSO (2.50 ppm), and 13C NMR chemical shifts were referenced to the solvent signal of DMSO (39.52 ppm).Fourier transform infrared spectroscopy (FTIR) was conducted with a Bruker Alpha-P ATR-spectrometer (Bruker, Ettlingen, Germany) in attenuated total reflection configuration. The data evaluation was carried out with the program OPUS (Bruker, Ettlingen, Germany).
Differential scanning calorimetry (DSC) was performed with a computer-controlled Phoenix DSC 204 F1 thermal analyzer (Netzsch, Selb, Germany). The samples were placed in aluminum pans (in air), which were cold-sealed and punctured. Before recording the DSC thermogram, the samples were cooled to −60 °C. Measurements were carried out at a heating rate of 5 °C min−1 under nitrogen flow (40 mL min−1). Given temperatures correspond to the onset of the respective thermal process. Given temperatures of glass transition correspond to the midpoint.
Thermogravimetric analysis (TGA) was performed with a TG 449 F3 Jupiter (Netzsch, Selb, Germany). Samples were placed under ambient atmosphere in aluminum oxide crucibles for the measurements. Measurements were carried out with a heating rate of 10 °C min−1 and air as the purge gas. Given temperatures correspond to a 5% onset.
Optical analyses were made by heated-stage polarized optical microscopy (POM) with an Axio Imager A1 microscope (Carl Zeiss MicroImaging GmbH, Göttingen, Germany) equipped with a hot stage, THMS600 (Linkam Scientific Instruments Ltd, Surrey, United Kingdom), and Linkam TMS 94 temperature controller (Linkam Scientific Instruments Ltd, Surrey, United Kingdom). Images were recorded at a magnification of 100× as a video with a digital camera. During heating and cooling, the sample was placed between two cover slips. Heating and cooling rates were 5 °C min−1.
A SYNAPT G2-S HDMS Q-ToF mass spectrometer (Waters, Manchester, United Kingdom), with an ESI operated in the positive and negative ion mode, was used in this study. For the measurement, the ion source was operated with a capillary voltage of 2500 V, extractor voltage: 1.0 V, RF lens: 0.5 V, ion source temperature of 120 °C, and desolvation temperature 250 °C. Nitrogen was used as both the cone and desolvation gas at a flow of 70 L h−1 and 500 L h−1, respectively. Argon was used as a collision gas at a pressure of 2.95 × 10−4 mbar.
Single crystal X-ray diffraction (SCXRD) data were collected on a Bruker Venture diffractometer (Bruxer AXS, Karlsruhe, Germany) using CuKα radiation (λ = 1.54178 Å at 100 K) or MoKα (λ = 0.71073 Å at 100 K). The crystal structures were solved and refined with SHELXT29 and SHELXL30 subroutines within the APEX331 software package. Absorption corrections were carried out with the program SADABS.32 Hydrogen atoms for all structures were calculated geometrically and treated with the riding atom mode. The positional disorder of the C12′ position (split) in 6 has been refined, yielding practically equivalent occupations (0.54/0.46(2)). Illustrations were prepared in Diamond33 and calculated diffractograms were obtained using Mercury.34
Powder X-ray diffraction (PXRD) data were recorded at ambient temperature on a PANalytical X'pert PRO diffractometer (Malvern Panalytical, Malvern, United Kingdom), operating at 45 kV and 40 mA and using CuKα1 radiation. The data were recorded in reflection mode from 5° to 70° with a step size of 0.01° for 60 min. The detector was a PIXcel3D (Malvern Panalytical, Malvern, United Kingdom) solid-state hybrid pixel detector.
Hirshfeld surfaces and images were calculated and produced using CrystalExplorer17.35
3. Results and discussion
3.1 Synthesis
 | ||
| Scheme 1 Synthetic procedures for 1–6. | ||
3.2 Thermal analyzes
 | ||
| Fig. 1 DSC traces for compounds 1–6. | ||
The DSC data indicate that all six salts meet the definition of an IL with melting points below 100 °C, ranging from the room temperature liquid 1 with Tg = −56.9 °C (Table 1) to Tm = 33.8–98.3 °C for 2–6. Crystallization of 1 could not be achieved, but an estimation according to Tm = 1.5 Tg (K)36 yields Tm = 51.2 °C. Upon cooling, 2 re-crystallizes, albeit with significant supercooling of 67.2 °C. The salts 3–5, once molten, do not crystallize upon cooling, but exhibit strong super cooling and vitrification way below room temperature – a thermal behavior, which is often found for ILs.36,37 Compound 6 with melting upon heating and crystallization upon cooling shows similar thermal behavior to compound 2, albeit with a much lower degree of supercooling (8.4 °C).
| Compound | Melting point (Tm, °C) | Crystallization (Tcryst, °C) | Glass transition (Tg, °C) | Decomposition temperature (T5%onset, °C) | |
|---|---|---|---|---|---|
| a Estimated according to Tm = 1.5 Tg (K). | |||||
| 1 | [Et3NH][MeTzS] | 51.2a | NA | −56.9 | 164 |
| 2 | [DBAH][MeTzS] | 97.0 | 29.8 | NA | 122 |
| 3 | [Et3NH][PhTzS] | 78.6 | NA | −40.2 | 126 |
| 4 | [DBAH][PhTzS] | 97.8 | NA | −15.0 | 127 |
| 5 | [Et3NH][MeDiTS] | 33.8 | NA | −40.8 | 164 |
| 6 | [DBAH][MeDiTS] | 98.3 | 89.9 | NA | 169 |
The thermal stabilities of the six salts were investigated by thermogravimetric analysis (TGA) from 25 °C to 500 °C under air with a thermal ramp of 10 °C min−1 (Fig. 2 and S25–S30†). The 5% onset of decomposition temperatures (T5%onset) are provided in Table 1. 1 slowly loses some mass almost immediately upon heating and exhibits T5%onset at 164 °C, degrading in one rather broad step ending at 332 °C. 2 is stable till 110 °C and loses almost 90% of its mass in a single step from 122 °C to 200 °C. A second mass loss starts at 210 °C until a total mass loss at 250 °C. Both 3 and 4 are stable until 110 °C, but then slowly decompose until 126 °C and 127 °C, respectively. From there, they both lose around 60% mass until 200 °C and then slowly continue their decomposition process until the end of the measurement, which could contain multiple decomposition steps, where less than 10% of the mass remains. 5 has a small mass loss at the beginning of the measurement, likely to be a minute amount of residual methanol, and then is stable until its decomposition temperature at 164 °C. In about 50 °C, the compound is fully decomposed. 6 decomposes in two well-defined steps, the first step at 82 °C and the second step at 169 °C. A third of the mass is lost in the first step, which can also be observed in DSC (for the first heating trace, the heating content is 6.26 J g−1, and for the second one, the value is 4.28 J g−1). After the second step, the compound is fully decomposed.
 | ||
| Fig. 2 TGA curves of 1–6. | ||
All the six salts have T5%onset values between 122 and 169 °C (Table 1). Interestingly, the only liquid, 1, has one of the highest T5%onset values of 164 °C, while 2, with one of the higher melting points (97.0 °C), has the lowest T5%onset of 122 °C. These two compounds have the same anion ([MeTzS]−), suggesting that, in this case, the cation has an important impact on the stability of the product. This contrasts with the thermal behavior of 3 and 4, which have the same anion ([PhTzS]−) with different cations, but the decomposition curves are very similar.
5 and 6, [MeDiTS]− salts, have different decomposition curves, but they are fully decomposed around the same temperature (210 °C), suggesting the cation/anion interactions play some role in the mechanism of decomposition. 6, with [DBAH]+ as the cation, exhibits a two-step decomposition (Fig. S30†), the first step roughly corresponding to the loss of the two isopropyl groups from the cation (32.7% mass loss (experimental) vs. 33.0% (theoretical) in the first step). The second step leads to complete decomposition.
The observed results cannot be directly related to the nature of the cations in this limited study. For the diprotic [DBAH]+ salts 2, 4, and 6, the melting points are similar (97.0–98.3 °C) but their solidification behavior is distinctly different. 2 shows large supercooling, 4 does not recrystallize at all, and 6 recrystallizes readily. In comparison, the [Et3NH]+ salt 1 is a liquid while 3 and 5 melt at lower temperatures (78.6 °C and 33.8 °C, respectively) and do not recrystallize under the experimental conditions, rather exhibiting glass transitions around −40.2 °C and −40.8 °C, respectively.
3.3 Crystal structures
SCXRD confirms full proton transfer from the thiol to the amine and formation of the respective ammonium cation and azole thiolate anion in each salt. For all solid compounds (2–6), crystals of sufficient quality for SCXRD could be grown by isothermal evaporation of solutions in either methanol (2–4) or ethyl acetate (for the [MeDiTS]-based compounds 5 and 6). Compounds 2–5 crystallize with one cation and one anion in the asymmetric unit and compound 6 – with two formula units. To fully assess any interactions that are important in the context of anti-crystal engineering, first, the structures of the individual ions as observed in the structures will be discussed in the context of the respective counterion, and then the crystal packing will be analyzed.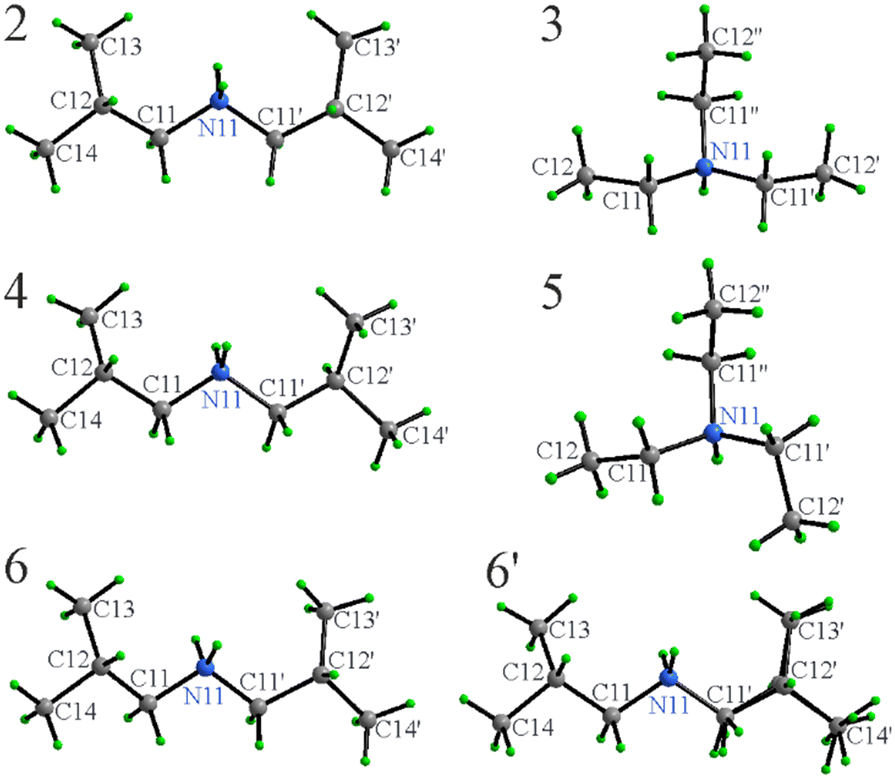 | ||
| Fig. 3 Molecular structures and atom label assignments for cations in 2–6. The second unique cation in the asymmetric unit of 6 (6′) is disordered (shown) with position C12′ split into two nearly identically occupied positions. Note the different conformations of the cations in 3 and 5. | ||
The conformationally flexible [Et3NH]+ cations do exhibit differences depending upon the anion or more precisely, their mutual orientation to optimize the packing (Fig. 3). The ethyl groups in 3 are T shaped with ∠C12N11C12′, ∠C12′′N11C12′ and ∠C12N11C12′′ of 92.2(1), 94.2(1), and 172.8(1)°, while in 5, the shape approaches an equilateral triangle with the angles 104.19(7), 125.13(7), and 119.19(7)°, respectively. [Et3NH]+ cations in both 3 and 5 are, in general, in similar conformations as one would expect from these types of molecules within the expected variations.38,40–42
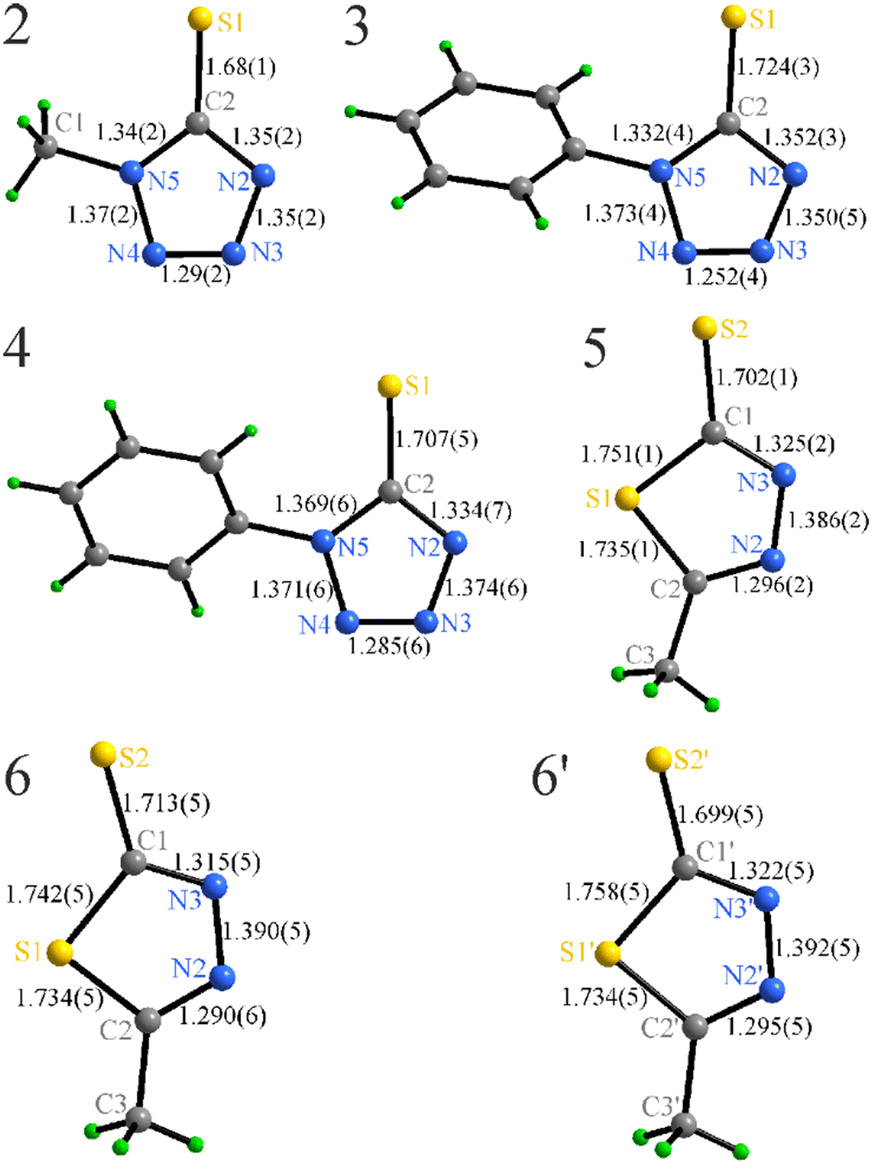 | ||
| Fig. 4 Bond lengths (Å) within the anions in 2–6. | ||
The phenyl groups in 3 and 4 are responsible for a slight distortion of the azole ring that can be followed by the C–N and N–N bond variations. The tetrazole and phenyl ring of the anion exhibit an angle between them, 47.7(1)° in 3 and 46.6(1)° in 4, showing a minor response to the cation variation.
The azole rings in 5–6 are significantly more distorted (the C–S–C angles in the latter are in the range 88.0–88.8(2)°) that is directly related to the incorporation of a larger S atom directly in the ring. These values are similar to the free 5-methyl-1,3,4-dithiazole-2-thiol (90.0°),48 or for example, in a mixed phenyl phosphate complex of CuI (90.2°).49
The anions in 2–4 appear to be moderately affected by both the type of cation and the R-group substitution, showing minor elongation of the C2–S1 bond, shortening of the N3–N4 contacts, and slight redistribution of the intra-ring angles (Fig. 4, Table S1†).
| Type | 2 | 3 | 4 | 5 | 6 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| d | ∠ | d | ∠ | d | ∠ | d | ∠ | d | ∠ | |
| N11(H)⋯N2 | 2.84(1) | 2.850(6) | ||||||||
| H⋯N2 | 1.93(1) | 1.979(4) | ||||||||
| ∠N11–H–N2 | 178.0(6) | 159.6(3) | ||||||||
| N11(H)⋯N3 | 2.95(1) | 2.738(2) | 2.782(5) | |||||||
| H⋯N3 | 2.04(1) | 1.820(2) | 1.872(4) | |||||||
| ∠N11–H–N3 | 177.6(7) | 174(2) | 177.9(3) | |||||||
| N11(H)⋯S1 | 3.265(3) | 3.296(4) | 3.257(4) | |||||||
| H⋯S1 | 2.540(3) | 2.432(1) | 2.386(1) | |||||||
| ∠N11–H–S1 | 149(3) | 158.6(2) | 150.4(2) | |||||||
| C11(H)⋯S2 | 3.601(5) | |||||||||
| H⋯S2 | 2.695(1) | |||||||||
| ∠C11–H–S2 | 152.3(3) | |||||||||
The [DBAH]+ salts 2, 4, and 6 present extended hydrogen-bonded motifs, including solely N–H⋯N connectivity in 2 and mixed N–H⋯N and N–H⋯S in 4 and 6 (Fig. 5). The presence of two N–H donors in this cation results in motifs that extend in space and are represented by zigzag chains in all structures with coplanar azole rings. Additionally, weaker C–H⋯S connectivity (dH–S = 2.695(1) Å) in 6 between the chains leads to the formation of formal layers, where azole rings in a given hydrogen-bonded chain belong to the same plane, while the azole rings in neighboring chains belong to parallel planes.
 | ||
| Fig. 5 Hydrogen-bonded (dotted blue) motifs in the crystal structures of 2, 4, and 6. | ||
The situation in the [Et3NH]+ salts 3 and 5 is different. The number of N–H donors is limited to one, and the formation of extended motifs is geometrically blocked, resulting in hydrogen bonded ion pairs with either weaker N–H⋯S (3, Fig. 6) or stronger N–H⋯N (5) interactions. In 3, the N–H⋯S hydrogen bond is of moderate strength at 2.540(3) Å,51,52 however, there are other significant interactions. Although π–π stacking is not observed, the C11–H⋯π and C12′′–H⋯π contacts are relatively short (2.72(1) and 2.82(1)) Å (Fig. 6), leading to the formation of a rather uniform network. This motif alternates 2-side C–H⋯π stacking and N–H⋯S hydrogen bonding, so each anion is connected to three cations while each cation connects to two anions.
 | ||
| Fig. 6 Hydrogen bonded (dotted blue) ionic pairs in the crystal structures of 3 and 5. C–H⋯π connectivity in 3 is indicated with dotted red lines. | ||
Since the positions of the cations in 3 and 5 are restricted due to interaction with the phenyl ring, it is not surprising that the thiol S atom wins the competition for the N–H-bond donor. Overall, the system sacrifices potentially stronger N–H⋯N connectivity towards the strength-wise uniform combination of N–H⋯S and C–H⋯π, finally resulting in a higher melting point. Interestingly, 5 also exhibits a similar layered structure, though, in contrast to 3, there are strong directional N–H⋯N bonds (1.820(2) Å). On the other hand, the C12–H⋯π in 5 connections involving the azole ring are slightly longer (weaker) (2.85–2.88(3) Å).
 | ||
| Fig. 7 2D fingerprints derived from the Hirshfeld surfaces showing the different types of intermolecular interactions dominating in each compound. | ||
It can be noted that the general shape of the fingerprints seems to be mainly dependent on the cation, indicating similar intermolecular interactions for similar cations. Another striking feature is the clear presence of the H⋯N interactions between the cation and the thiol ring (characterized by sharp spikes denoted with a red star) in all molecules except of 3. One should also note that the H⋯S interactions (denoted with a green star) appear to be more prominent in 3, 4, and 6 with no direct relation to the type of the cation. In addition, and expectedly, H⋯C interactions are well represented in the two compounds containing [PhTzS]− (3–4) and [MeDiTS]− (5–6) as they exhibit noticeable C–H⋯π interactions (characterized by a “wing” in the fingerprint).
 | ||
| Fig. 8 Projection of a single layer in the crystal structures of 2 on the bc plane (a) and the crystal packing on the ac plane (b). Apolar segments have been highlighted. Crystallographic axes are color-coded: a = red, b = green, c = blue. | ||
 | ||
| Fig. 9 Projection of a single layer in the crystal structures of 3 and 5 on the ab plane (a and c, respectively) and the crystal packing on the bc plane (b and d, respectively). Crystallographic axes are color-coded: a = red, b = green, c = blue. | ||
 | ||
| Fig. 10 Projection of a single layer in the crystal structures of 4 on the bc plane (a), of 6 on the ac plane (b) and the crystal packing of 6 on the ab plane (c). Crystallographic axes are color-coded: a = red, b = green, c = blue. | ||
All three diprotic [DBAH]+ salts (2, 4, and 6) with two charge assisted N–H hydrogen bonds exhibit seemingly more ordered structures. The variety of hydrogen-bond interactions further translates into the packing diagram of the ILs, with similar zigzag patterns being exhibited according to the cation present in the molecule. Given their broad connectivity, it is understandable that these have the highest and similar melting points (all above 95 °C) of the salts studied here.
The decomposition temperatures seem to be dependent on the anion's identity rather than the connectivity between the ion pairs. In fact, both compounds 3 and 4 and compounds 5 and 6, respectively, composed of [PhTzS]− and [MeDiTS]− exhibit decomposition temperatures within a couple of degrees from each other (126 ± 1 °C for 3 and 4 and 166 ± 3 °C for 5 and 6). However, the highest decomposition temperatures are also exhibited by the compounds displaying the strongest contribution of the N–H⋯S bond.
Conclusions
Six new ILs, including one room temperature IL, containing both N- and S-donors, have been synthesized and characterized. The crystal structures of the five crystalline solids reveal extensive hydrogen bonded networks that are cation dependent. Three of the compounds (3, 4, and 6) present relatively rare charge assisted N–H⋯S hydrogen bonds. While C–H⋯π interactions are present in nearly all these salts, it plays a supportive role in those with strong N–H⋯S hydrogen bonding. By contrast, C–H⋯π interactions appear to be the main driving force in the thermal behavior of 3. Although hydrogen bonding is important in these salts, there does not seem to be one type, which can be assigned as the major driver for the observed physical properties of a given IL, but rather the combination of multiple intermolecular interactions. These dissimilarities can easily be observed from the Hirshfeld surface fingerprints between 3 and 5, directly pointing to different connectivities in the solid state.The thermal behavior of [DBAH]+ salts is strongly dictated by polymeric hydrogen bonded motifs leading to higher melting points, while [Et3NH]+ salts allow stronger competition between different types of bonding and show higher differentiation of their melting temperatures. Even the limited set of investigated compounds suggest that control of the hydrogen bond network could be a tool for designing the ILs' properties. Future research will be focused on the correlation between the specific structural features, hydrogen bonding, and the resulting physico-chemical characteristics to allow fine tuning towards desired properties and will consequently lead to further expansion of the library of task specific ILs.
Author contributions
OR, GB: methodology, investigation, writing. VS: analysis, writing, visualization. AVM, RDR: conceptualization, supervision, analysis, writing, funding support.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
A.-V. M. would like to thank the Royal Academy of Sciences, Sweden, for support through the Göran Gustafsson prize in Chemistry, Energimydigheten (The Swedish Energy Agency) for support through grant no. 46676-1 and Stiftelsen för Strategisk Forskning (Swedish Foundation for Strategic Research, SSF) for support in the REFIT research consortium. A.-V. M. and R. D. R. acknowledge the Swedish Research Council for a Tage Erlander professorship to R. D. R. (VR grant 2018-00233). R. D. R would like to thank the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Heavy Elements program for support under award DE-SC0019220.References
- L. Mei, P. Ren, Q.-Y. Wu, Y.-B. Ke, J.-S. Geng, K. Liu, X.-Q. Xing, Z.-W. Huang, K.-Q. Hu, Y.-L. Liu, L.-Y. Yuan, G. Mo, Z.-H. Wu, J. K. Gibson, Z.-F. Chai and W.-Q. Shi, J. Am. Chem. Soc., 2020, 142, 16538–16545 CrossRef CAS PubMed.
- F. W. Lewis, L. M. Harwood, M. J. Hudson, M. G. B. Drew, J. F. Desreux, G. Vidick, N. Bouslimani, G. Modolo, A. Wilden, M. Sypula, T.-H. Vu and J.-P. Simonin, J. Am. Chem. Soc., 2011, 133, 13093–13102 CrossRef CAS PubMed.
- C. Xiao, Z. Hassanzadeh Fard, D. Sarma, T.-B. Song, C. Xu and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 16494–16497 CrossRef CAS PubMed.
- D. Prodius, M. Klocke, V. Smetana, T. Alammar, M. Perez Garcia, T. L. Windus, I. C. Nlebedim and A.-V. Mudring, Chem. Commun., 2020, 56, 11386–11389 RSC.
- D. Prodius, K. Gandha, A.-V. Mudring and I. C. Nlebedim, ACS Sustainable Chem. Eng., 2020, 8, 1455–1463 CrossRef CAS.
- A. V. Rudnev, Russ. Chem. Rev., 2020, 89, 1463 CrossRef CAS.
- J.-C. G. Bünzli, Trends Chem., 2019, 1, 751–762 CrossRef.
- D. Prodius and A.-V. Mudring, Coord. Chem. Rev., 2018, 363, 1–16 CrossRef CAS.
- X. Sun, H. Luo and S. Dai, Chem. Rev., 2012, 112, 2100–2128 CrossRef CAS.
- A. P. Abbott, G. Frisch, J. Hartley and K. S. Ryder, Green Chem., 2011, 13, 471–481 RSC.
- I. Billard, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, 2013, vol. 43, pp. 213–273 Search PubMed.
- A. Rout and K. Binnemans, Dalton Trans., 2014, 43, 3186–3195 RSC.
- M. P. Jensen and A. H. Bond, J. Am. Chem. Soc., 2002, 124, 9870–9877 CrossRef CAS PubMed.
- A. Bhattacharyya, P. Mohapatra and V. Manchanda, J. Radioanal. Nucl. Chem., 2011, 288, 709–716 CrossRef CAS.
- V. A. Cocalia, M. P. Jensen, J. D. Holbrey, S. K. Spear, D. C. Stepinski and R. D. Rogers, Dalton Trans., 2005, 1966–1971 RSC.
- S. P. Kelley, J. S. Nuss and R. D. Rogers, in Application of Ionic Liquids on Rare Earth Green Separation and Utilization, ed. J. Chen, Springer, Heidelberg, 2016, pp. 21–42 Search PubMed.
- X. Han and D. W. Armstrong, Acc. Chem. Res., 2007, 40, 1079–1086 CrossRef CAS PubMed.
- L. Xu, N. Pu, Y. Li, P. Wei, T. Sun, C. Xiao, J. Chen and C. Xu, Inorg. Chem., 2019, 58, 4420–4430 CrossRef CAS PubMed.
- S. Laufer, G. Wagner and D. Kotschenreuther, Angew. Chem., Int. Ed., 2002, 41, 2290–2293 CrossRef CAS PubMed.
- M. E. Easton, H. Choudhary and R. D. Rogers, Chem. – Eur. J., 2019, 25, 2127–2140 CrossRef CAS PubMed.
- D. M. S. Paqhaleh, L. Hashemi, V. Amani, A. Morsali and A. Aminjanov, Inorg. Chim. Acta, 2013, 407, 1–6 CrossRef CAS.
- M. Taheriha, M. Ghadermazi and V. Amani, J. Mol. Struct., 2016, 1107, 57–65 CrossRef CAS.
- W. Lu, P. S. Barber, S. P. Kelley and R. D. Rogers, Dalton Trans., 2013, 42, 12908–12916 RSC.
- A. Marakushev and N. Bezmen, Int. Geol. Rev., 1971, 13, 1781–1794 CrossRef.
- J. S. Preston and A. C. D. Preez, Solvent Extr. Ion Exch., 1994, 12, 667–685 CrossRef CAS.
- T. Vander Hoogerstraete, B. Onghena and K. Binnemans, J. Phys. Chem. Lett., 2013, 4, 1659–1663 CrossRef.
- D. Yaprak, E. T. Spielberg, T. Bäcker, M. Richter, B. Mallick, A. Klein and A.-V. Mudring, Chem. – Eur. J., 2014, 20, 6482–6493 CrossRef CAS.
- P. M. Dean, J. Turanjanin, M. Yoshizawa-Fujita, D. R. MacFarlane and J. L. Scott, Cryst. Growth Des., 2009, 9, 1137–1145 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- Bruker AXS Inc., Wisconsin, USA, 2016.
- L. Krause, R. Herbst-Irmer and D. Stalke, J. Appl. Crystallogr., 2015, 48, 1907–1913 CrossRef CAS.
- DIAMOND: Program for Crystal and Molecular Structure Visualization, Crystal Impact GbR, Bonn, Germany, 2011 Search PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- S. Wolff, D. Grimwood, J. McKinnon, M. Turner, D. Jayatilaka and M. Spackman, Crystal explorer, 2012 Search PubMed.
- A.-V. Mudring, Aust. J. Chem., 2010, 63, 544–564 CrossRef CAS.
- O. Renier, G. Bousrez, M. Yang, M. Hölter, B. Mallick, V. Smetana and A.-V. Mudring, CrystEngComm, 2021, 23, 1785–1795 RSC.
- A. Piecha-Bisiorek, A. Białońska, R. Jakubas, P. Zieliński, M. Wojciechowska and M. Gałązka, Adv. Mater., 2015, 27, 5023–5027 CrossRef CAS PubMed.
- A. Suvitha, B. Varghese and M. N. Sudheendra Rao, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o344–o346 CrossRef CAS.
- L. E. Shmukler, I. V. Fedorova, M. S. Gruzdev and L. P. Safonova, J. Phys. Chem. B, 2019, 123, 10794–10806 CrossRef CAS.
- A. V. Churakov and J. A. Howard, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2004, 60, o557–o558 CrossRef.
- T. J. Bednarchuk, V. Kinzhybalo, E. Markiewicz, B. Hilczer and A. Pietraszko, J. Solid State Chem., 2018, 258, 753–761 CrossRef CAS.
- G. Liu and S. Zhang, Z. Kristallogr. - New Cryst. Struct., 2016, 231, 479–480 CrossRef CAS.
- O. Jiménez-Sandoval, R. Cea-Olivares and S. Hernández-Ortega, Polyhedron, 1997, 16, 4129–4135 CrossRef.
- M. Hernández-Arganis, R. A. Toscano, M. Moya-Cabrera, V. García-Montalvo and R. Cea-Olivares, Z. Anorg. Allg. Chem., 2004, 630, 1627–1631 CrossRef.
- K. Ortner and U. Abram, Polyhedron, 1999, 18, 749–754 CrossRef CAS.
- R. Cea-Olivares, O. Jiménez-Sandoval, S. Hernández-Ortega, M. Sánchez, R. A. Toscano and I. Haiduc, Heteroat. Chem., 1995, 6, 89–97 CrossRef CAS.
- F. Hipler, M. Winter and R. A. Fischer, J. Mol. Struct., 2003, 658, 179–191 CrossRef CAS.
- V. Stylidou, K. Kavaratzi, I. Papazoglou, A. G. Hatzidimitriou, A. G. Papadopoulos, P. Angaridis and P. Aslanidis, Eur. J. Inorg. Chem., 2018, 2018, 2915–2926 CrossRef CAS.
- P. A. Hunt, C. R. Ashworth and R. P. Matthews, Chem. Soc. Rev., 2015, 44, 1257–1288 RSC.
- L. A. H. van Bergen, M. Alonso, A. Palló, L. Nilsson, F. De Proft and J. Messens, Sci. Rep., 2016, 6, 30369 CrossRef CAS PubMed.
- P. Zhou, F. Tian, F. Lv and Z. Shang, Proteins: Struct., Funct., Bioinf., 2009, 76, 151–163 CrossRef CAS.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed.
- A. Parkin, G. Barr, W. Dong, C. J. Gilmore, D. Jayatilaka, J. J. McKinnon, M. A. Spackman and C. C. Wilson, CrystEngComm, 2007, 9, 648–652 RSC.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC.
- H. S. Biswal and S. Wategaonkar, J. Phys. Chem. A, 2009, 113, 12763–12773 CrossRef CAS PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, instrumentation, 1H and 13C NMR spectra, FTIR spectra, TG and DSC curves, PXRD diffractograms, SCXRD details, and POM images. CCDC 2084096–2084100 contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce00961g |
| This journal is © The Royal Society of Chemistry 2023 |