
Efficient end-group functionalization and diblock copolymer synthesis via Au(III) polymer reagents†
Grace E.
Kunkel
 a,
Joseph W.
Treacy
a,
Hayden R.
Montgomery
a,
Ellie G.
Puente
a,
Evan A.
Doud
a,
Alexander M.
Spokoyny
ab and
Heather D.
Maynard
*ab
a,
Joseph W.
Treacy
a,
Hayden R.
Montgomery
a,
Ellie G.
Puente
a,
Evan A.
Doud
a,
Alexander M.
Spokoyny
ab and
Heather D.
Maynard
*ab
aDepartment of Chemistry and Biochemistry, University of California, Los Angeles, 607 Charles E. Young Drive East, Los Angeles, California 90095-1569, USA. E-mail: maynard@chem.ucla.edu
bCalifornia NanoSystems Institute, University of California, Los Angeles, 570 Westwood Plaza, Los Angeles, California 90095-1569, USA
First published on 30th November 2023
Abstract
Herein, we describe the synthesis of bench-stable organometallic Au(III) terminated polymer reagents. These reagents mediate the chemoselective S-arylation of thiol-containing small molecules and polymers to yield functionalized mono-telechelic polymers and diblock copolymers, respectively. These transformations proceed rapidly within minutes and produce conjugates in quantitative conversion, making this strategy a robust addition to the polymer functionalization toolbox.
Polymers with α- and/or ω-functionalization, also known as telechelic polymers, are useful building blocks for the synthesis of unique macromolecular architectures through coordination and conjugation.1 This type of functionality can be achieved through the post-polymerization modification of chain transfer agents (CTAs) or use of functionalized CTAs in reversible addition fragmentation chain transfer (RAFT) polymerization, the nucleophilic substitution of terminal halides in atom transfer radical polymerization (ATRP) or the use of functionalized initiators, or the synthesis of nucleophilic initiators in anionic ring opening polymerization (ROP).1 For example, small molecules such as fluorescent probes and affinity tags are commonly conjugated to polymers post-synthetically for use in various biological and materials applications. While there are many successful examples of these strategies, the post-polymerization modification of the end-group can suffer from challenges including poor kinetics which results in moderate to low levels of conversion to product. Further, the resulting linkages can also be reversible or cleavable, leading to loss of the desired functionality.2
In addition to appending small molecules, telechelic polymers are also well-suited for the synthesis of macromolecular architectures such as diblock copolymers. Typically, synthesis of diblock copolymers is undertaken by sequential polymerization of different monomers.3 However, when the target diblock copolymer contains units not polymerizable by compatible methods, post-polymerization conjugation of the disparate polymer blocks is required.3 This latter synthetic strategy has significant challenges including the necessity of a highly efficient conjugation due to the low concentration of reactive units and the steric hindrance caused by polymer chains. One method to alleviate these synthetic concerns is to use “click”-type reactions due to their enhanced kinetics and chemoselectivity.4 Effective examples of using “click”-type reactions for the construction of diblock copolymers include thiol–ene reactions,5,6 Cu(I)-catalyzed azide–alkyne cycloadditions (CuAAC),7–9 Diels–Alder cycloadditions,10,11 and more recently developed selective routes such as Sulfur(VI) fluoride exchange (SuFEx) reactions.12 Thus, “click”-type chemistry allows for the facile synthesis of these diblock copolymers that otherwise would be synthetically inaccessible.11,13–16 In each case, careful design and successful installation of reactive polymer end-groups is critical to achieve the desired product. Despite enabling impressive macromolecular structures, it is important to note that traditional “click”-chemical routes face some limitations. For example, these aforementioned methods have been known to exhibit low conversion when coupling partners are used at equimolar ratios due to kinetic limitations.17 These methods can also place restrictions on monomer scope such as the need for the repeat units to be thermal- or photo-stable.18 “Click”-type reactions can also lack certain chemical orthogonality; for example, acetylenic Glaser coupling is a possible side reaction for CuAAC conjugations.19 Alternatively, the termination of living polymerizations with macromolecules has been utilized for the synthesis of these architectures, but this strategy generally requires a large excess of the terminating macromolecule, necessitating purification via time-intensive fractionation.20 There remains a need for additional rapid and mild methodologies to address these limitations in existing conjugation techniques.
We have previously developed Au(III) mediated S-arylation utilizing isolable and bench-stable (Me-DalPhos)Au(III)Aryl reagents.21 Recently, this chemistry has been expanded to demonstrate sterically-driven regioselectivity as well as successful picomolar bioconjugation with reagents enabling bimolecular rate constants of up to 1.7 × 104 M−1 s−1.22 Organometallic S-arylation with Au(III) oxidative addition complexes (OACs) are highly chemoselective, pH tolerant, and rapid at room temperature.21–23 With these characteristics in mind, we envisioned that Au(III) OACs would efficiently facilitate the synthesis of both mono-telechelic polymers and diblock copolymers via ligand exchange with a second thiol-containing species and subsequent reductive elimination (RE). The resulting S-aryl bond would obviate the concern of a reversible conjugation, and the rapid kinetics and chemoselectivity of the reaction would provide quantiative conversion at equimolar ratios and prevent undesired side reactions.
We first synthesized aryl iodide-capped polymers to serve as precursors to OACs (Fig. 1A). We prepared p(ε-caprolactone) (pCL) (1) (Fig. 1B) by anionic ROP using an aryl iodide-functionalized initiator (2) and the 3-O/MTBD cocatalyst sytem.24 While in preliminary polymerizations we utilized a 4-iodobenzyl alcohol to initiate, we found that upon oxidative addition, the terminal ester adjacent to the Au(III) complex can be activated and cleaved. Therefore, we replaced the initiator with 4-iodophenethyl alcohol (2) wherein the terminal ester was no longer activated at the benzylic position, and thus less likely to cleave. Next, p(n-butylnorborneneimide) (pBNI) (3) (Fig. 1C) was synthesized by ROMP and quenched using a cis-stilbene aryl iodide derivative (4) to incorporate an aryl iodide via direct end-capping.25,26 Finally, we synthesized an aryl iodide-containing ATRP initiator (5) from 2 and employed it in the synthesis of p(pentafluorostyrene) (pPFS) (6) (Fig. 1D). Many polymer conjugation strategies utilize the tertiary bromide of ATRP polymer end-groups,27 which generally necessitates low polymer conversion to protect end-group fidelity.28,29 In this case, the use of an aryl iodide-containing ATRP initiator ensures the presence of a functional handle without sacrificing polymer conversion. All aryl iodide polymers underwent oxidative addition in open air with (Me-DalPhos)Au(I)Cl (7) using AgSbF6 as a halide scavenger to afford isolable and bench stable Au(III) polymer reagents (1a, 3a, 6a). 1H and 31P{1H} NMR spectroscopy were used to determine conversion to the Au(III) species, and it was found that the removal of excess Au(I) was not necessary, as it did not inhibit the subsequent S-arylation. Isolated Au(III) polymer complexes are stable for up to at least three months, as monitored by 31P{1H} NMR spectroscopy.30
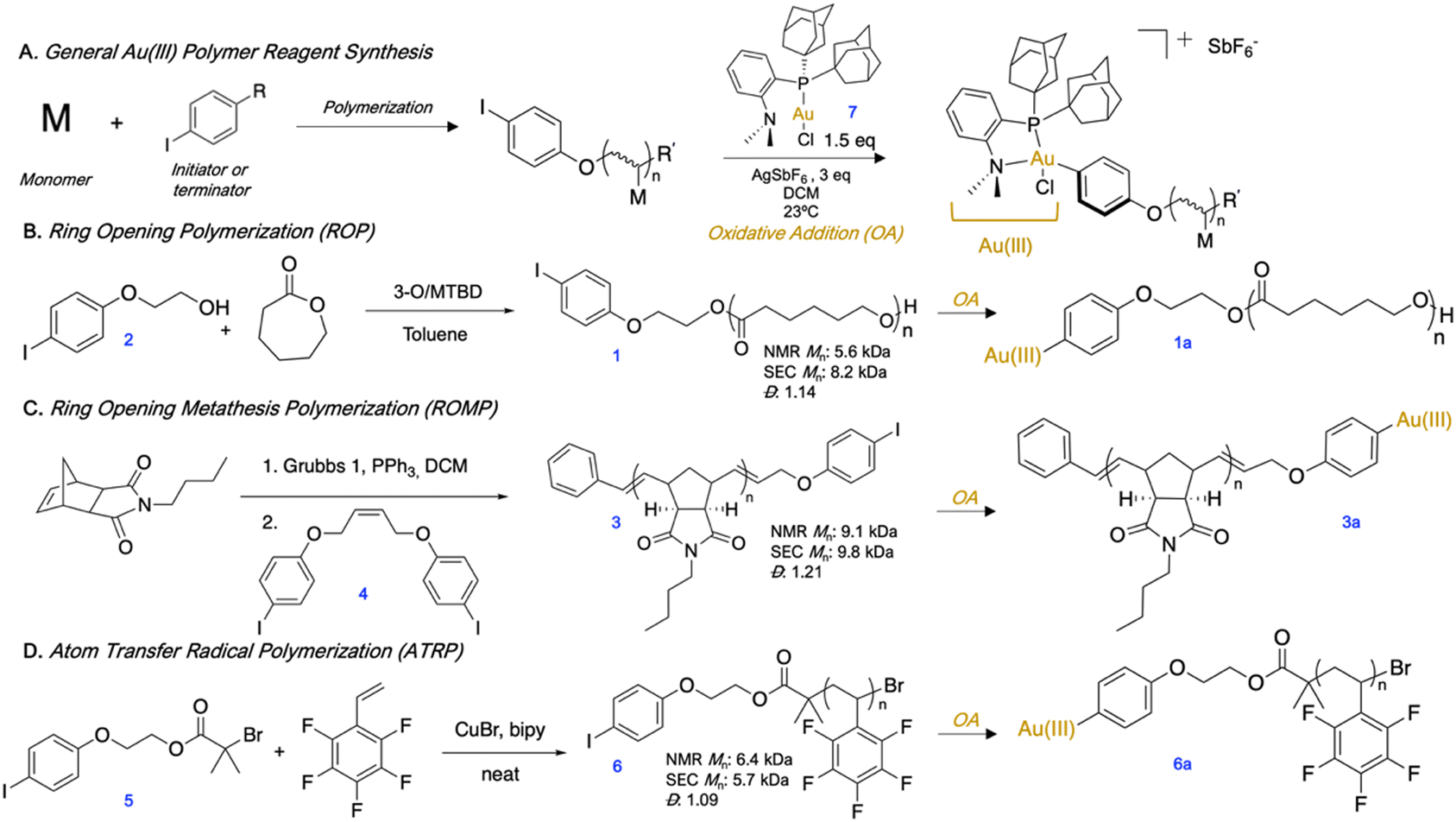 | ||
| Fig. 1 (A) General Au(III) polymer reagent synthesis (Au(III) = [(Me-DalPhos)Au(III)Cl]+ SbF6−). (B) Ring opening polymerization of ε-caprolactone and synthesis of OAC. (C) Ring opening metathesis polymerization of n-butylnorborneneimide and synthesis of OAC. (D) Atom transfer radical polymerization of pentafluorostyrene and synthesis of OAC. | ||
We performed S-arylation on a small library of biologically relevant small molecules (Fig. 2A). Successful S-arylation of thiolated biotin (8a), thiolated coumarin (9a), and commercial sodium thioglucose (TG) with pCL-Au(III) (1a) and pBNI-Au(III) (3a) occurred in 30 minutes as observed via1H NMR and 31P{1H} NMR spectroscopy (ESI,† Fig. S48–S60). MALDI-TOF characterization was also used to observe mass differences which correspond to the replacement of the iodide of 1a with 8a, thereby confirming that efficient S-arylation had occurred (Fig. 2B).
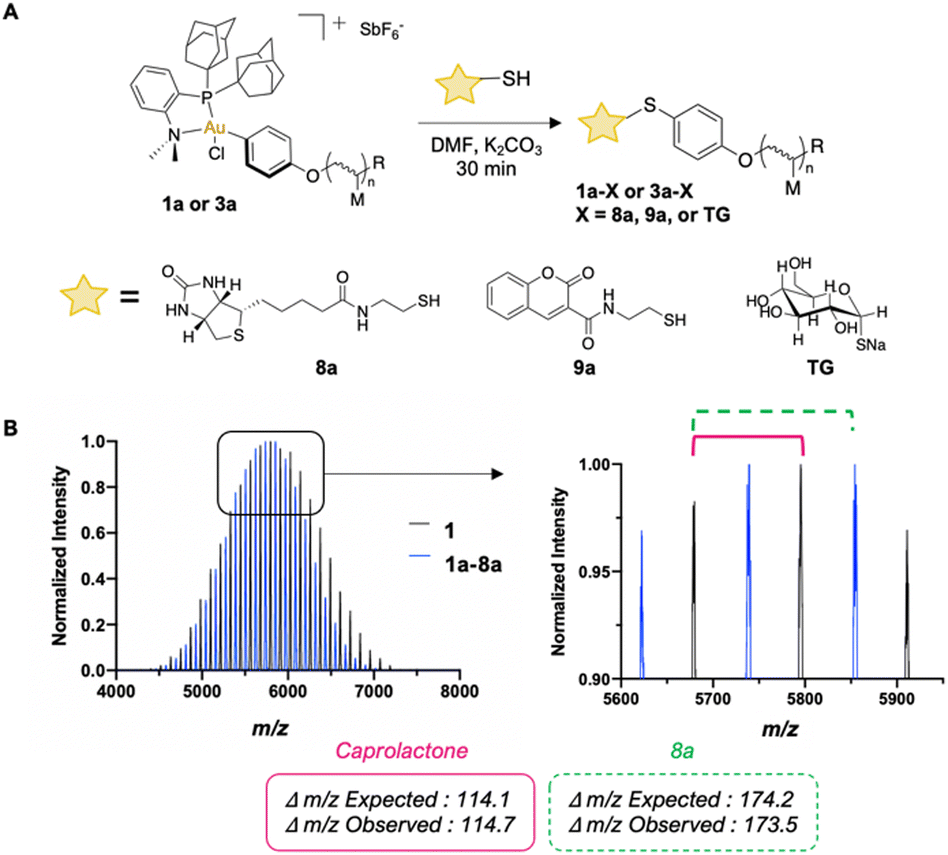 | ||
| Fig. 2 (A) Scheme of modified mono-telechelic polymer synthesis via S-arylation of thiolated biotin (8a), thiolated coumarin (9a), and sodium thioglucose (TG) with 1a or 3a. (B) MALDI-TOF mass spectra of 1 and 1a–8a with magnified inset. Expected and calculated Δm/z differences between repeat units of 1a and end groups of 1a and 1a–8a. | ||
In a similar approach, we hypothesized that these Au(III) polymer reagents would offer a facile and modular synthesis of diblock copolymers utilizing thiol-modified mono-telechelic polymers (Table 1). To this end, p(N-isopropylacrylamide) (pNIPAM) (10) was synthesized via RAFT. The presence of a dithioester in many CTAs affords a free thiol coupling partner following aminolysis (10a). This aminolysis of the CTA was monitored by UV-Vis spectroscopy and 1H NMR (ESI,† Fig. S63 and S64). To achieve thiol-functionalized polymers via ROP, an S-trityl protected thioether initiator (11) was used for the synthesis of pCL (12) and subsequently deprotected to reveal the free thiol (12a). This demonstrates that controlled polymerization strategies such as ROP can be utilized in either the thiol or aryl iodide block interchangeably by employing the appropriate small molecule conjugation handle in the initiator.
| Entry | Functionality | Precursor | Strategy | Expected Mn (kDa) | SEC Mn (kDa) | Dispersity (Đ) |
|---|---|---|---|---|---|---|
| 10 | Thiol | NIPAM | RAFT | 9.6 | 9.3 | 1.11 |
| 12 | Thiol | CL | ROP | 9.6 | 10.0 | 1.20 |
| 13 | BCP | 1a + 10a | S-Arylation | 12.5 | 11.9 | 1.37 |
| 14 | BCP | 3a + 12a | S-Arylation | 25.7 | 17.7 | 1.27 |
| 15 | BCP | 6a + 12a | S-Arylation | 12.5 | 11.9 | 1.37 |
| 16 | Aryl I | BNI | ROMP | 30.2 | 27.2 | 1.27 |
| 17 | Thiol | CL | ROP | 36.4 | 37.4 | 1.13 |
| 18 | BCP | 16a + 17a | S-Arylation | 72.7 | 42.3 | 1.40 |
This thiol polymer library was subjected to various polymeric Au(III) S-arylation reagents to yield diblock copolymers synthesized by disparate polymerization methods (Table 1, entries 13–15, see ESI† for synthetic details). Specifically, p(NIPAM)-SH (10a) was reacted with pCL-Au(III) (1a) to prepare a p(NIPAM)-b-p(CL) diblock copolymer (13). Furthermore, p(CL)-SH (12a) was reacted with p(BNI)-Au(III) (3a) and p(PFS)-Au(III) (6a) to produce p(CL)-b-p(BNI) (14) and p(CL)-b-p(PFS) (15), respectively. All S-arylation reactions occurred in one hour, in open air, using an equimolar ratio of polymer precursors, and at ambient temperature, highlighting the mild conditions and efficiency of this synthetic strategy. These reactions occurred in the presence of tributylphosphine (PBu3) as a disulfide reducing agent, and it was observed that PBu3 did not interfere with the S-arylation. Conversion and product dispersity were monitored by multinuclear NMR spectroscopy, diffusion ordered spectroscopy (DOSY), and size exclusion chromatography (SEC) (ESI,† Fig. S71–S87). DOSY NMR experiments indicated that, in every example, both sets of polymer peaks diffused at the same rate, suggesting one, connected diblock copolymer species in solution. Complete conversion of precursors 6a and 12a to produce 15 was observed by DOSY NMR experiments (ESI,† Fig. S86), and 19F NMR spectroscopy (ESI,† Fig. S83) indicates that the S-arylation outpaces any potential SNAr reactions with the side chains of 6a despite the lower relative concentration of Au(III) in solution.31 Since 1H, 31P{1H}, and DOSY NMR experiments indicated full conversion to diblock copolymer products, peak shape abnormalities in SEC spectra for S-arylation products may be a result of secondary structure and column interaction from disparately hydrophobic blocks.
In general, quantitative conversion to diblock copolymer products becomes more challenging as the polymer precursor size increases, as this lowers the relative concentration of reactive end-group units in solution. We hypothesized that this robust conjugation method would allow for access to large diblock copolymers that may be challenging to obtain using other methods. To test this hypothesis, 27.2 kDa pBNI-Au(III) (16a) and 36.4 kDa pCL-SH (17a) mono-telechelic polymers were prepared (Table 1). We observed quantitative conversion to diblock copolymer product 18 by 31P{1H} and DOSY NMR spectroscopy after one hour using our standard conjugation conditions, highlighting the efficiency of this method (Fig. 3).
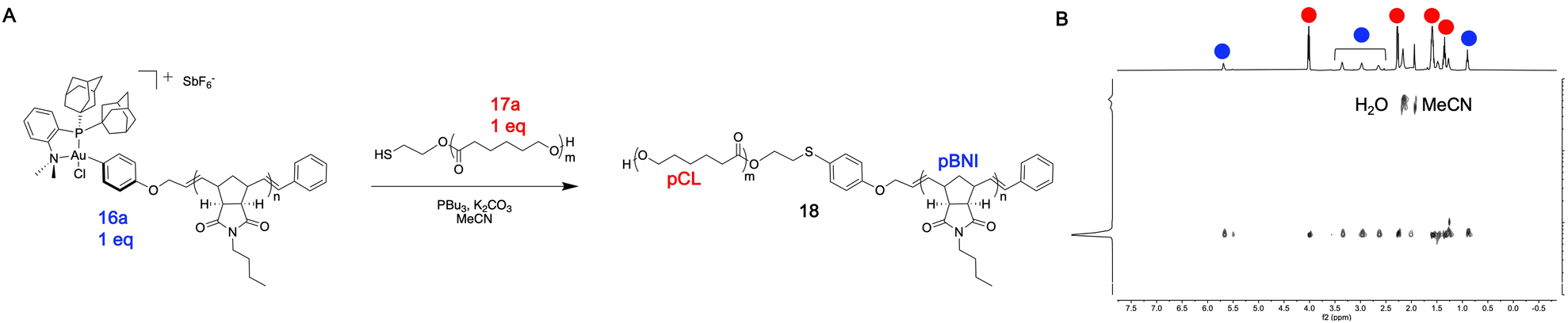 | ||
| Fig. 3 (A) pCL-b-pBNI (18) S-arylation scheme (B) DOSY NMR spectrum of pCL-b-pBNI (18) in CD3CN. | ||
This work demonstrates the efficiency of the post-polymerization synthesis of small molecule mono-telechelic polymers and diblock copolymers utilizing organometallic Au(III) polymer reagents. The synthetic availability of the aryl iodide and thiol coupling partners allows for their facile incorporation into small molecules and polymers. These polymers can be synthesized by common controlled polymerization techniques such as RAFT, ROP, ATRP, and ROMP. The selectivity of (Me-DalPhos)Au(I)Cl (7) for aryl iodides during oxidative addition and the thiophilicty of Au(III) permits the use of many desirable side-chain functional groups without concern of cross-reactivity. Au(III) polymer reagents are isolable and bench-stable, allowing for a modular approach to the rapid minute-scale synthesis of various functionalized polymers. Ultimately, this work adds to the “click”-type reaction toolbox for the synthesis of complex polymers and can be expanded to the preparation of other polymeric applications and macromolecular architectures.
G. E. K., J. W. T., H. R. M., E. A. D., A. M. S., and H. D. M. conceived and designed experiments. G. E. K. and J. W. T. performed the majority of synthesis. G. E. K., J. W. T., H. R. M., and E. G. P. analyzed the data. G. E. K. wrote the original manuscript.
This work was funded by the NSF (CHE-2003946). GEK was supported by the NIH under award number T32GM008496. These studies were supported by the NSF instrumentation grant CHE-1048804. A. M. S. thanks NIGMS (R35GM124746) for funding.
Conflicts of interest
There are no conflicts to declare.References
- J. Kim, H. Y. Jung and M. J. Park, Macromolecules, 2020, 53, 746–763 CrossRef CAS.
- S. N. S. Alconcel, S. H. Kim, L. Tao and H. D. Maynard, Macromol. Rapid Commun., 2013, 34, 983–989 CrossRef CAS.
- H. Dau, G. R. Jones, E. Tsogtgerel, D. Nguyen, A. Keyes, Y.-S. Liu, H. Rauf, E. Ordonez, V. Puchelle, H. Basbug Alhan, C. Zhao and E. Harth, Chem. Rev., 2022, 122, 14471–14553 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- J. M. Spruell, B. A. Levy, A. Sutherland, W. R. Dichtel, J. Y. Cheng, J. F. Stoddart and A. Nelson, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 346–356 CrossRef CAS.
- M. Plank, F. V. Frieß, C. V. Bitsch, J. Pieschel, J. Reitenbach and M. Gallei, Macromolecules, 2023, 56, 1674–1687 CrossRef CAS.
- J. A. Opsteen and J. C. M. van Hest, Chem. Commun., 2005, 57–59 RSC.
- W. Agut, D. Taton and S. Lecommandoux, Macromolecules, 2007, 40, 5653–5661 CrossRef CAS.
- N. Toncheva-Moncheva, P. Bakardzhiev, S. Rangelov, B. Trzebicka, A. Forys and P. D. Petrov, Macromolecules, 2019, 52, 3435–3447 CrossRef CAS.
- H. Durmaz, A. Dag, O. Altintas, T. Erdogan, G. Hizal and U. Tunca, Macromolecules, 2007, 40, 191–198 CrossRef CAS.
- A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2009, 48, 2411–2414 CrossRef CAS.
- J. C. Brendel, L. Martin, J. Zhang and S. Perrier, Polym. Chem., 2017, 8, 7475–7485 RSC.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC.
- Z. Geng, J. J. Shin, Y. Xi and C. J. Hawker, J. Polym. Sci., 2021, 59, 963–1042 CrossRef CAS.
- A. Shahrokhinia, P. Biswas and J. F. Reuther, J. Polym. Sci., 2021, 59, 1748–1786 CrossRef CAS.
- S. P. S. Koo, M. M. Stamenović, R. A. Prasath, A. J. Inglis, F. E. Du Prez, C. Barner-Kowollik, W. Van Camp and T. Junker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1699–1713 CrossRef CAS.
- H. Durmaz, B. Colakoglu, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1667–1675 CrossRef CAS.
- C. J. Duxbury, D. Cummins and A. Heise, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3795–3802 CrossRef CAS.
- J. M. Notestein, L.-B. W. Lee and R. A. Register, Macromolecules, 2002, 35, 1985–1987 CrossRef CAS.
- M. S. Messina, J. M. Stauber, M. A. Waddington, A. L. Rheingold, H. D. Maynard and A. M. Spokoyny, J. Am. Chem. Soc., 2018, 140, 7065–7069 CrossRef CAS.
- E. Doud, J. Tilden, J. Treacy, E. Chao, H. Montgomery, G. Kunkel, N. Adhami, T. Kerr, A. Rheingold, C. Frost, K. Houk, H. Maynard and A. Spokoyny, ChemRxiv, 2023, preprint DOI:10.26434/chemrxiv-2023-brjnz.
- J. A. R. Tilden, A. T. Lubben, S. B. Reeksting, G. Kociok-Köhn and C. G. Frost, Chem. – Eur. J., 2022, 28, e202104385 CrossRef CAS.
- K. V. Fastnacht, S. S. Spink, N. U. Dharmaratne, J. U. Pothupitiya, P. P. Datta, E. T. Kiesewetter and M. K. Kiesewetter, ACS Macro Lett., 2016, 5, 982–986 CrossRef CAS.
- S. Hilf, R. H. Grubbs and A. F. M. Kilbinger, J. Am. Chem. Soc., 2008, 130, 11040–11048 CrossRef CAS PubMed.
- A. E. Madkour, A. H. R. Koch, K. Lienkamp and G. N. Tew, Macromolecules, 2010, 43, 4557–4561 CrossRef CAS PubMed.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS.
- N. V. Tsarevsky, B. S. Sumerlin and K. Matyjaszewski, Macromolecules, 2005, 38, 3558–3561 CrossRef CAS.
- M. Zhong and K. Matyjaszewski, Macromolecules, 2011, 44, 2668–2677 CrossRef CAS.
- H. R. Montgomery, M. S. Messina, E. A. Doud, A. M. Spokoyny and H. D. Maynard, Bioconjugate Chem., 2022, 33, 1536–1542 CrossRef CAS PubMed.
- C. R. Becer, K. Babiuch, D. Pilz, S. Hornig, T. Heinze, M. Gottschaldt and U. S. Schubert, Macromolecules, 2009, 42, 2387–2394 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc05350d |
| This journal is © The Royal Society of Chemistry 2024 |