
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisualizing molecular deformation in fibrin networks under tensile loading via FLIM–FRET†
Mohammadhasan
Hedayati‡
 a,
Yuan-I
Chen‡
a,
Justin R.
Houser
a,
Yujen
Wang
a,
Sajjad
Norouzi
a,
Hsin-Chih
Yeh
ab and
Sapun H.
Parekh
*a
a,
Yuan-I
Chen‡
a,
Justin R.
Houser
a,
Yujen
Wang
a,
Sajjad
Norouzi
a,
Hsin-Chih
Yeh
ab and
Sapun H.
Parekh
*a
aBiomedical Engineering, University of Texas at Austin, Austin, TX, USA. E-mail: sparekh@utexas.edu
bTexas Materials Institute, University of Texas at Austin, Austin, TX, USA
First published on 9th November 2023
Abstract
Mapping molecular deformation and forces in protein biomaterials is critical to understanding mechanochemistry. Here we use intramolecular Förster resonance energy transfer (FRET) of dual-labeled fibrin to distinguish molecular conformations of proteins in situ during mechanical loading. The FRET approach offers increased spatial resolution compared to our previous vibrational imaging. By using fluorescence lifetime microscopy (FLIM), we demonstrate that the combination of FRET and FLIM can probe the molecular changes in fibrin with high spatial (nanometer) and temporal (nanosecond) resolution. Our results map changes in fibrin monomer deformation during the macroscopic loading of the fibrin network, paving the way to directly visualizing the biomaterial mechanics and structure in cell-ECM scaffolds for the first time.
Fibrin plays an essential role in haemostasis, both as the main structural element of blood clots and as the scaffold for cell recruitment in wound healing.1 At sites of vascular injury, fibrin forms from its precursor, fibrinogen (FIB), which is converted to fibrin via thrombin cleavage.2 Fibrin monomers subsequently polymerize into protofibrils and eventually form a crosslinked network of fibers that supports platelet adhesion and aggregation and entraps red blood cells.3,4 During wound healing, the resulting fibrin network must be strong enough to withstand the shear and tensile forces from blood flow while being flexible enough to accommodate large strains from muscle contraction.5 The biophysical properties of fibrin networks are critical factors that determine the functionality of the blood clot and also minimize the risks of premature clot rupture and embolism. For instance, fibrin fibers under external mechanical loads exhibit different orientations, thicknesses, and molecular structures, which can ultimately regulate the downstream biological readouts at the enzymatic and cellular levels.6–8 Cellular receptors and enzymes that interact with fibrin do so via molecular-scale contacts. However, the mechanisms connecting physical and molecular changes in fibrin and downstream biochemical signaling are unclear. Thus, a major research challenge remains in understanding how mechanical forces impact the molecular structure of fibrin proteins.
Förster resonance energy transfer (FRET) between fluorophores attached to proteins provides an in situ tool to study molecular distances, conformation of proteins, and molecular forces for various applications.9 FRET is a physical process by which energy is transferred by dipole–dipole coupling from an excited state donor fluorophore to a ground state acceptor when appropriate spectral overlap and proximity requirements are satisfied.10 FRET-based measurements can be used to resolve intra- and intermolecular distances on the nanometer length scale within heterogeneous biopolymers such as fibrin fibers. This technique offers the physical measurement of molecular distances and forces in response to (macroscopic or microscopic) external loading. In this study, we have used the FRET technique to quantify the molecular deformation of fibrin molecules within a fibrin network as a function of macroscopic applied strains.
Inspired by work from Vogel and colleagues on fibrinonectin,11–13 FIB was dual-labelled with donor (Atto 488) and acceptor (Alexa 568) fluorophores based on NHS ester reactivity toward amine groups that are available in FIB (Fig. 1). Fig. S1 (ESI†) shows possible labelling locations, with a concentration in the β and γ nodule domains. Atto 488, with an emission peak at 520 nm, and Alexa 568, with an excitation peak at 579 nm were selected as the donor and acceptor for FRET. The integral overlap between the fluorescence emission spectrum of Atto 488 and the fluorescence excitation spectrum of Alexa 568 is high enough to induce dipole–dipole coupling (Fig. 1). Monomers were dual labelled with a high density of acceptors and low density of donors, such that the average donor is substantially quenched by the surrounding pool of acceptors. This allows, in principle, for a high FRET efficiency between the donor and acceptor on a FIB molecule in a fibrin network. We initially measured the FRET efficiency of dual-labelled fibrin (DL FIB) as a probe within a fibrin network through acceptor photobleaching. We empirically determined that a 10% DL FIB + 90% unlabelled FIB mixture achieved the appropriate balance between the fluorescence signal for the microscope while minimizing the number of modified monomers in the fibrin network. In acceptor photobleaching, the intensity of the donor is enhanced by the depletion of the acceptor via photo-destruction. Fig. S2a (ESI†) demonstrates that the average intensity of fibrin fibers within the designated area of the donor channel is considerably higher after photobleaching of the acceptor dye, proving the presence of FRET. We also observed that by increasing the molar ratio of the acceptor to donor from 1.5 to 4.5 (1.5, 2.5, 3.4, 4.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the FRET efficiency increases; however, for ratios greater than 3.4, we did not observe a significant difference in FRET efficiency, which indicates that the number of acceptors on the FIB molecules had saturated. For all subsequent experimental studies, we chose the ratio of 3.4:1 as a compromise between strong FRET and efficient use of dye molecules. To further clarify that FRET was due to intramolecular dual-labelling, we also formed fibrin gels from the combination of pure FIB with 10% of singly-labelled FIB Atto 488 and singly-labelled FIB Alexa 568 (Fig. S2b, ESI†). The combination of singly-labelled FIB Atto 488 with FIB Alexa 568 showed a FRET efficiency of ∼5%, which is substantially lower than the average of ∼48% efficiency for the fibrin gels formed from 10% DL FIB + 90% unlabelled FIB. This indicates that the majority of our FRET signal comes from intramolecular FRET, which provides a distance between the donor and the acceptor molecules that is shorter than the Förster distance (<∼6 nm).
:1), the FRET efficiency increases; however, for ratios greater than 3.4, we did not observe a significant difference in FRET efficiency, which indicates that the number of acceptors on the FIB molecules had saturated. For all subsequent experimental studies, we chose the ratio of 3.4:1 as a compromise between strong FRET and efficient use of dye molecules. To further clarify that FRET was due to intramolecular dual-labelling, we also formed fibrin gels from the combination of pure FIB with 10% of singly-labelled FIB Atto 488 and singly-labelled FIB Alexa 568 (Fig. S2b, ESI†). The combination of singly-labelled FIB Atto 488 with FIB Alexa 568 showed a FRET efficiency of ∼5%, which is substantially lower than the average of ∼48% efficiency for the fibrin gels formed from 10% DL FIB + 90% unlabelled FIB. This indicates that the majority of our FRET signal comes from intramolecular FRET, which provides a distance between the donor and the acceptor molecules that is shorter than the Förster distance (<∼6 nm).
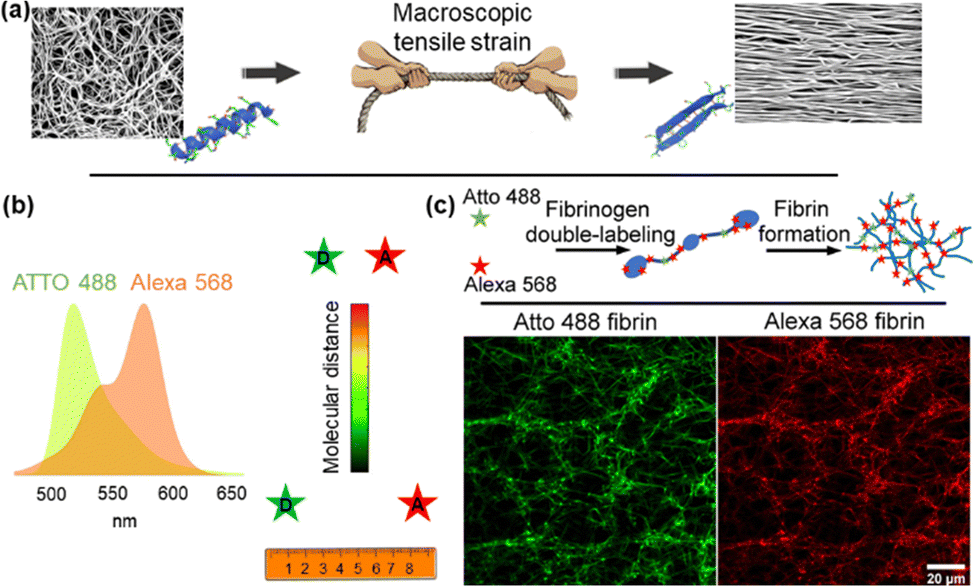 | ||
| Fig. 1 Quantitative measurement of intramolecular distance as a function of external loads in a dual-labelled fibrin network with the FRET method. (a) External forces applied to fibrin cause the structural transitions from α-helices to β-sheets. (b) The spectrum of Atto 488's emission overlaps with the excitation spectrum of Alexa 568. When they are in proximity (less than ∼10 nm), the donor (Atto 488) and acceptor (Alexa 568) exhibit FRET. (c) FIB dual labelling with Atto 488 as a donor and Alexa 568 as an acceptor and the use of this probe in fibrin networks. | ||
With DL FIB as a FRET sensor, we used fluorescence lifetime imaging (FLIM) to measure quantitative FRET between acceptor and donor dyes, in a non-destructive way within at-rest and strained fibrin networks.14 Moreover, because fiber packing increases with tensile strain due to volume conservation and water expulsion,6 the fibrin density in the field of view increases strongly, skewing intensity-based FRET measurements. To measure FLIM–FRET in situ, we used a simple tensile loading device (Fig. S3, ESI†) to apply strains on the microscope and measure the donor lifetime of Atto 488 in the fibrin network at increasing strains. Upon stretching the fibrin networks to 100% and 160% tensile strain, we observed significant fibrin fiber alignment along the loading direction in the XY plane (Fig. 2a). Large tensile deformation in fibrin has been shown to cause secondary structural changes in fibrin;7,15 however, no studies have experimentally correlated these structural changes with physical deformations in the FIB molecule.
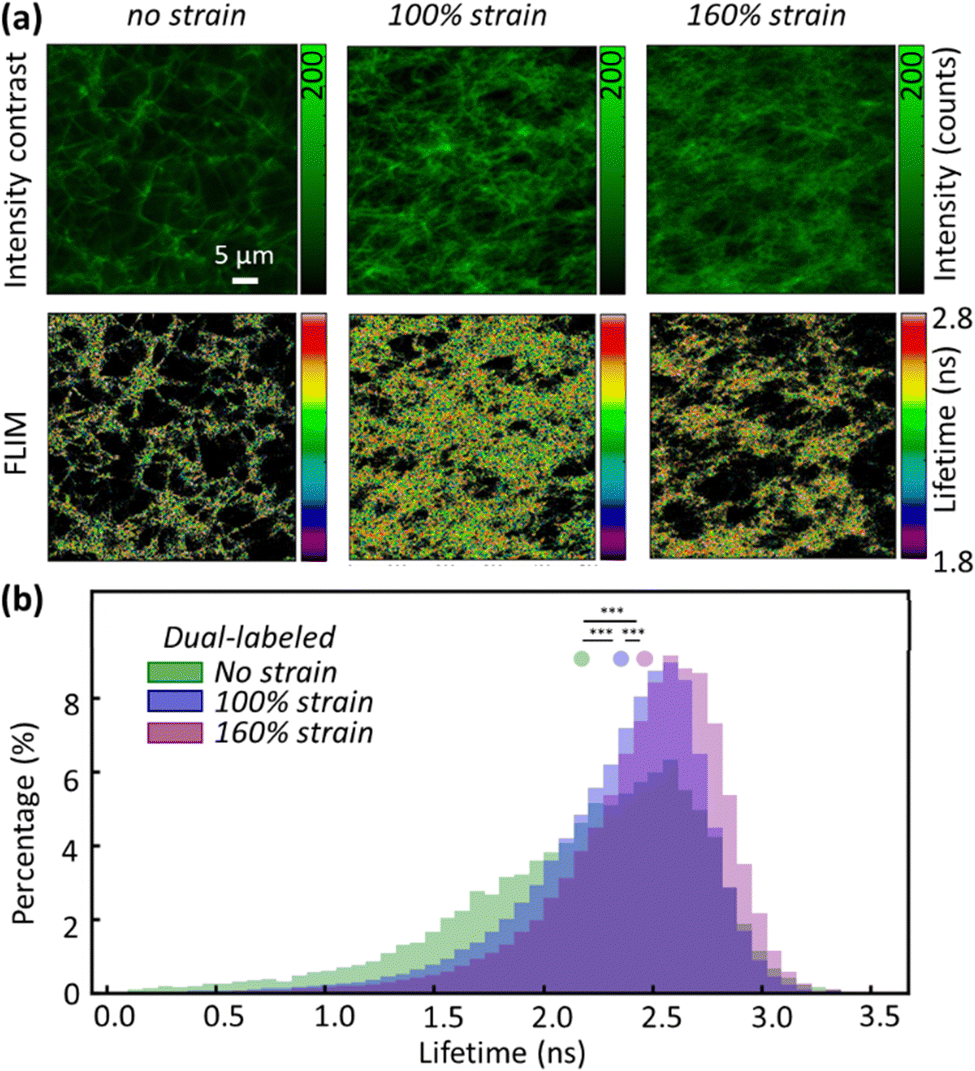 | ||
| Fig. 2 Stretching the fibrin network decreases the FRET efficiency. (a) Intensity contrast images (top) and FLIM images (bottom) obtained from 0%, 100%, and 160% tensile strain show an increasing trend of mean lifetime in response to larger tensile strain. (b) Normalized lifetime histograms obtained from 0%, 100%, and 160% tensile strain show a rise in fluorescence lifetime because of tensile strain. Significant differences (***p < 0.001) are shown in between using the KS (Kolmogorov–Smirnov) test. | ||
In the absence of an acceptor, FLIM images of fibrin showed a mean lifetime of 3.22 ± 0.18 ns (mean ± standard deviation, s.d.), near the expected lifetime of Atto 488 in solution. In contrast, the mean lifetime of DL FIB in unloaded fibrin fibers was 2.12 ± 0.65 ns (Fig. 2b). This agrees with our acceptor photobleaching results showing that the Atto 488 is quenched by the acceptor (Alexa 568), upon network formation. Applying 100% and 160% tensile strain resulted in decreased FRET and a corresponding increase of donor lifetime. The increase in lifetime of the donor dye suggests nanometer-scale deformations in the FIB molecule, which increased the distance between donor and acceptor fluorophores and decreased FRET. At 100% and 160% tensile strain, we observed 9% and 15% longer mean lifetime values (2.32 ± 0.47 ns and 2.43 ± 0.45 ns for 100% and 160% tensile strain), respectively. At strains below 100%, we did not observe a significant increase in donor lifetime. This can be attributed to the minimal alteration in the donor–acceptor distance or excessive quenching of the donor by acceptor saturation, which would consequently limit the impact on the donor lifetime.
In addition to increased donor lifetime, we observed a narrower distribution of lifetime values when the fibrin was stretched, indicating that the initially heterogeneous, unstrained fibrin network responded to the tensile strain such that a larger percentage of fibers exhibited a more homogenous loading (Fig. 2b and Fig. S4, ESI†). As the donor lifetime changed in DL FIB under strain, the corresponding energy transfer efficiency (E): E = 1 − (τDA/τD) (where τDA and τD are the donor lifetimes in the presence and absence of acceptor) for 100% and 160% strain decreased by 18% and 26%, respectively (E = 0.34, 0.28, 0.25 for 0%, 100%, and 160% tensile strain, Fig. 3a). These demonstrate the expected trend of decreasing FRET at higher tensile strain. A normalized, stacked graph was used to show the heterogenous composition analysis of lifetime in FLIM images and illustrate how the contributions of subcategories change with different strains (Fig. 3b). The lifetime shorter than 2.2 ns (high FRET efficiency subcategories) constituted 45%, 30%, and 21% of unstretched, and 100% and 160% stretched fibrin, respectively. In contrast, more than 60% of total pixels at 100% strain showed lifetime values ranging from 2.2 ns to 2.8 ns, demonstrating the decrease of the FRET efficiency (as compared with 47% at 0% strain). Interestingly, we observed a 2.14-fold increase of the longer lifetime's subcategory contribution (>2.8 ns) from 100% to 160% strain, where the coiled coils in fibrin molecules are known to undergo secondary structural transitions.8
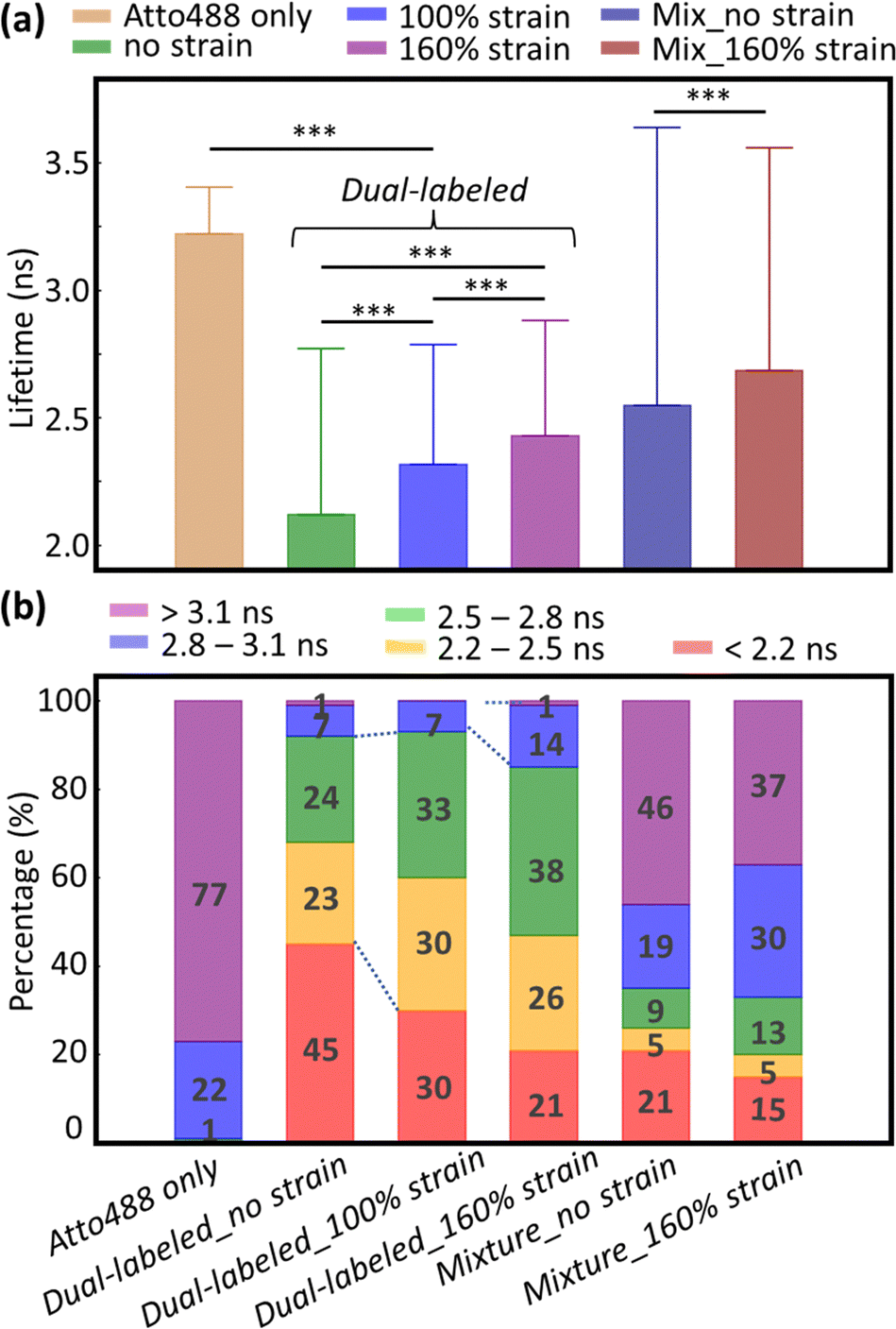 | ||
| Fig. 3 Characterizing the tensile strain effect on the FRET efficiency. (a) Bar plot shows the mean fluorescence lifetime for each condition (error bars, s.d.). All the pairs exhibit significant differences (***p < 0.001 using a two-sided paired t-test). (b) Stacked histogram of the fluorescence lifetime distribution for each condition. Percentages of each subcategory are displayed in the figure. | ||
A second observation is the alignment of fibrin fibers with load and the relation with donor lifetime (Fig. 4a and b). We used OrientationJ to estimate the local predominant orientation (see ESI†).16 The orientational density of local angles has been plotted in both rectangular and polar coordinates, showing the expected increased alignment of fibers in the axial direction (at 0°) by 45% and 55% for 100% and 160% strain (Fig. 4b). Fig. 2 also indicates that the unstretched fibrin has well-separated fibers distributed isotropically. The distribution of lifetimes with fiber orientation was broader before the tensile strain was applied to the network. An integrative analysis of fiber orientation and donor lifetime provided evidence for the fibrin strain-bearing mechanism (Fig. 4c). When applying 100% tensile strain on the unstretched fibrin, a visible alignment in orientation analysis and a narrower lifetime distribution with a greater mean value appeared. This likely results from stretching of the disordered αC domains that straighten at much lower strains, as well as secondary structural changes in the coiled coil region (closer to 100% strain).7,17 Further increasing the tensile strain from 100% to 160% unfolds the coiled coils and β and γ nodule domains in fibrin.18 At a strain of 160%, the change in fiber orientation was minimal but the increase of fibrin's molecular deformation could be monitored via the increasing fluorescence lifetime (Fig. 4c). Fig. S5 (ESI†) depicts a larger field of view of donor lifetime at different macroscopic strains, showing no obvious spatially dependent FRET in the network. Our findings are consistent with acceptor and donor dyes being attached to stiffer nodules or coiled coil domains rather than flexible αC domains; based on the amino acid composition (Fig. S1, ESI†), we believe that the dyes are located in the nodules.
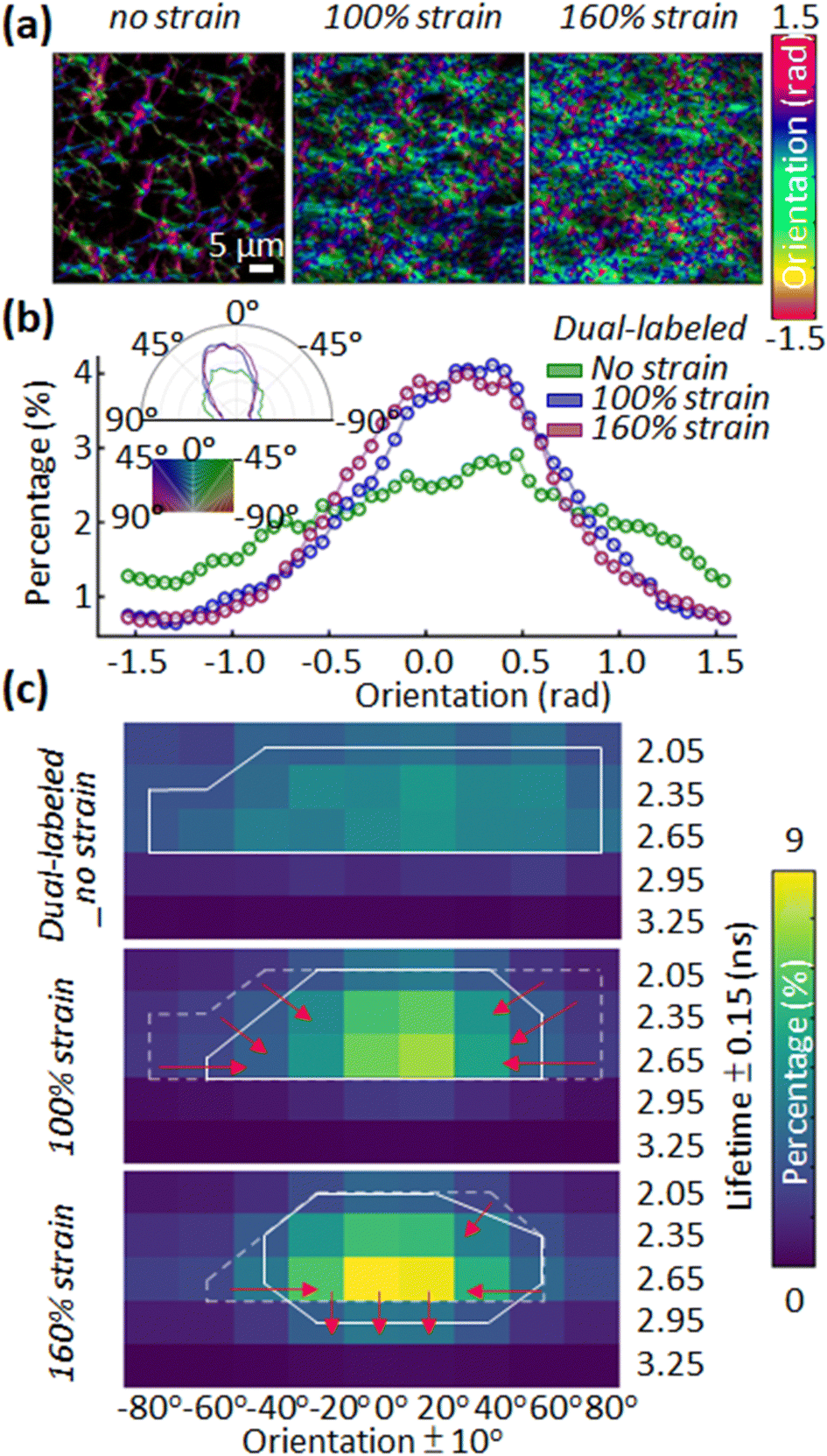 | ||
| Fig. 4 Characterizing the effect of tensile strain on the local orientation angle and FIB deformation. (a) Orientation maps of the fibrin network obtained from dual-labelled 0%, 100%, and 160% tensile strains show an increasing alignment in response to larger tensile strain. (b) Histogram of XY fiber orientation angles at increasing tensile strain. Inset, polar histogram of the XY fiber orientation angles. (c) A heatmap of the 2D histogram of orientation and fluorescence lifetime was obtained from 0%, 100%, and 160% tensile strain. Each pixel in the histogram represents the percentage of the number of pixels in the image showing a fluorescence lifetime and an orientation within certain ranges. The solid white line is a contour enclosing the region with a percentage greater than 2%. Dotted white lines are the enclosing region from the previous strain for comparison. | ||
In our previous work, we reported secondary structure transitions in fibrin at 100% strain using BCARS.7 These transitions can be correlated with strain-dependent molecular deformations observed in FRET measurements here. The proximity of donors and acceptors on fibrinogen monomers leads to intramolecular FRET. Donors and acceptors are close enough to induce a shorter mean donor lifetime in the initially formed fibrin fibers. However, only after sufficient macroscopic tensile strain is applied, the alignment, and more importantly, stretching (unfolding) of fibrin increase the donor–acceptor distance, resulting in increased donor lifetime and decreased FRET efficiency.19 Interestingly, we observe a finite FRET efficiency even after 160% strain, which indicates either incomplete protein unfolding or a limitation of the FRET sensor's sensitivity. Protein unfolding could be suppressed by protofibrils sliding with respect to one another.6 Additionally, the dyes may be localized such that they are sensitive to molecular deformation of only a certain region of the protein. We aim to further explore the dye positioning on fibrinogen to optimize the sensitivity of this deformation sensor.
Previous studies have unveiled the biochemistry and mechanical behaviour of fibrin fibers in networks. These investigations have provided insights into changes in fiber alignment, packing, and secondary structure though with limited imaging capability.7,17 By using FLIM–FRET, we can map the impact of macroscopic loading on the molecular deformation of fibrin monomers in the network in a facile way. With the ability to visualize molecular deformation with fiber-scale spatial resolution of FLIM–FRET, we hope to relate molecular deformation and strains in whole blood clots.
M. H. and S. H. P. conceived the idea; M. H., Y.-I. C., and S. H. P designed the study. M. H., J. R. H., Y. W., and S. N. prepared the samples and built the loading stage. M. H. and Y.-I. C. performed the FRET experiments. Y.-I. C. supported the instrumental setups and Y.-I. C. developed image processing and analysis methods. H.-C. Y. and S. H. P. supervised the study. M. H. and Y.-I. C. drafted the manuscript. All authors edited and finalized the manuscript.
We acknowledge Jeanne Stachowiak and Carl Hayden for helpful discussions about this work. This work was supported by a National Science Foundation grant to S. H. P. (2105175) and a Welch Foundation grant to S. H. P. (F-2008-20220331). The support to Y.-I. C. and H.-C. Y. is from the National Science Foundation (2029266), National Institutes of Health (EY033106) and Texas Global Faculty Research Steed Grant.
Conflicts of interest
There authors have no conflicts to declare.References
- J. W. Weisel and R. I. Litvinov, Blood, 2013, 121, 1712–1719 CrossRef CAS.
- M. Hedayati, M. J. Neufeld, M. M. Reynolds and M. J. Kipper, Mater. Sci. Eng., R, 2019, 138, 118–152 CrossRef.
- M. Hedayati, M. M. Reynolds, D. Krapf and M. J. Kipper, ACS Appl. Mater. Interfaces, 2018, 10, 31892–31902 CrossRef CAS.
- J. R. Vlcek, M. Hedayati, A. C. Melvin, M. M. Reynolds and M. J. Kipper, Adv. Healthcare Mater., 2021, 10, 2001748 CrossRef CAS.
- J. W. Weisel, Biophys. Chem., 2004, 112, 267–276 CrossRef CAS.
- A. E. X. Brown, R. I. Litvinov, D. E. Discher, P. K. Purohit and J. W. Weisel, Science, 2009, 325, 741–744 CrossRef CAS.
- Y. Wang, S. Kumar, A. Nisar, M. Bonn, M. K. Rausch and S. H. Parekh, Acta Biomater., 2021, 121, 383–392 CrossRef CAS.
- S. Kumar, Y. Wang, M. Hedayati, F. Fleissner, M. K. Rausch and S. H. Parekh, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2117675119 CrossRef CAS.
- C. Grashoff, B. D. Hoffman, M. D. Brenner, R. Zhou, M. Parsons, M. T. Yang, M. A. McLean, S. G. Sligar, C. S. Chen, T. Ha and M. A. Schwartz, Nature, 2010, 466, 263–266 CrossRef CAS.
- T. Chen, B. He, J. Tao, Y. He, H. Deng, X. Wang and Y. Zheng, Adv. Drug Delivery Rev., 2019, 143, 177–205 CrossRef CAS.
- G. Baneyx, L. Baugh and V. Vogel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5139–5143 CrossRef CAS.
- E. Klotzsch, M. L. Smith, K. E. Kubow, S. Muntwyler, W. C. Little, F. Beyeler, D. Gourdon, B. J. Nelson and V. Vogel, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18267–18272 CrossRef CAS.
- M. L. Smith, D. Gourdon, W. C. Little, K. E. Kubow, R. A. Eguiluz, S. Luna-Morris and V. Vogel, PLoS Biol., 2007, 5, e268 CrossRef.
- S. Ranjit, L. Malacrida, D. M. Jameson and E. Gratton, Nat. Protoc., 2018, 13, 1979–2004 CrossRef CAS.
- F. Fleissner, M. Bonn and S. H. Parekh, Sci. Adv., 2016, 2, e1501778 CrossRef.
- Z. Püspöki, M. Storath, D. Sage and M. Unser, in Focus on Bio-Image Informatics, ed. W. H. De Vos, S. Munck and J.-P. Timmermans, Springer International Publishing, Cham, 2016, vol. 219, pp. 69–93 Search PubMed.
- B. E. Vos, C. Martinez-Torres, F. Burla, J. W. Weisel and G. H. Koenderink, Acta Biomater., 2020, 104, 39–52 CrossRef CAS.
- A. Zhmurov, O. Kononova, R. I. Litvinov, R. I. Dima, V. Barsegov and J. W. Weisel, J. Am. Chem. Soc., 2012, 134, 20396–20402 CrossRef CAS PubMed.
- G. Baneyx, L. Baugh and V. Vogel, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14464–14468 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed methods and supplementary data. See DOI: https://doi.org/10.1039/d3cc05281h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |