
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnlocking the reactivity of diazo compounds in red light with the use of photochemical tools†
Katarzyna
Orłowska
,
Klaudia
Łuczak
,
Piotr
Krajewski
 ,
João V.
Santiago
,
Katarzyna
Rybicka-Jasińska
* and
Dorota
Gryko
*
,
João V.
Santiago
,
Katarzyna
Rybicka-Jasińska
* and
Dorota
Gryko
*
Institute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52 01-224, Warsaw, Poland. E-mail: dorota.gryko@icho.edu.pl
First published on 22nd November 2023
Abstract
Structurally diversified diazoalkanes can be activated under red light irradiation relying on direct photolysis, photosensitization or photoredox catalysis.
Bioorthogonal chemistry represents chemical transformations that proceed selectively in biological environments without perturbing the structure of biomolecules or interfering with biochemical pathways.1–6 Several photoactivated methods have been designed, yet most of them employ short-wavelength light emitting sources.7–10 The phototoxicity of high energetic photons makes them inappropriate for biological applications. Therefore, switching to less energetic light is desirable. Along this line, tetrazole bioorthogonal chemistry was performed under NIR radiation via two-photon excitation or upconversion processes.11,12 Dihydrotetrazine oxidation in vivo can also be achieved with less energetic photons.13 Yet, red light-induced reactions even in synthetic chemistry call for in depth studies.14
Diazoalkanes are versatile reactants for photochemical synthesis of small/complex structures15–17 and functionalization of bioactive compounds.18–21 They have been utilized in enzymatic cyclopropanation, ring expansion, cyclopropenation, or insertion reactions.22–27 So far, however, generation of carbenes in biological systems is mostly limited to diazirines that are activated in UV/violet light.28–30 In view of benefits arising from the application of low energetic photons, red light-induced diazo chemistry is highly desirable. Given the structural diversity of diazoalkanes, they can be directly photolyzed or activated via photocatalytic processes under visible light (even red, Fig. 1A). We wondered whether it is possible to unlock the potential of red light toward the generation of reactive species from structurally diversified diazo compounds utilizing various photochemical modes. While studying the photocatalytic activity of porphyrins under red-light irradiation, we found that they catalyze photoalkylation of aldehydes with ethyl diazoacetate.31 Herein, we present our comprehensive study on the red light-induced photolysis, photosensitization, and photoredox-driven generation of reactive intermediates from diazo reagents (Fig. 1B).
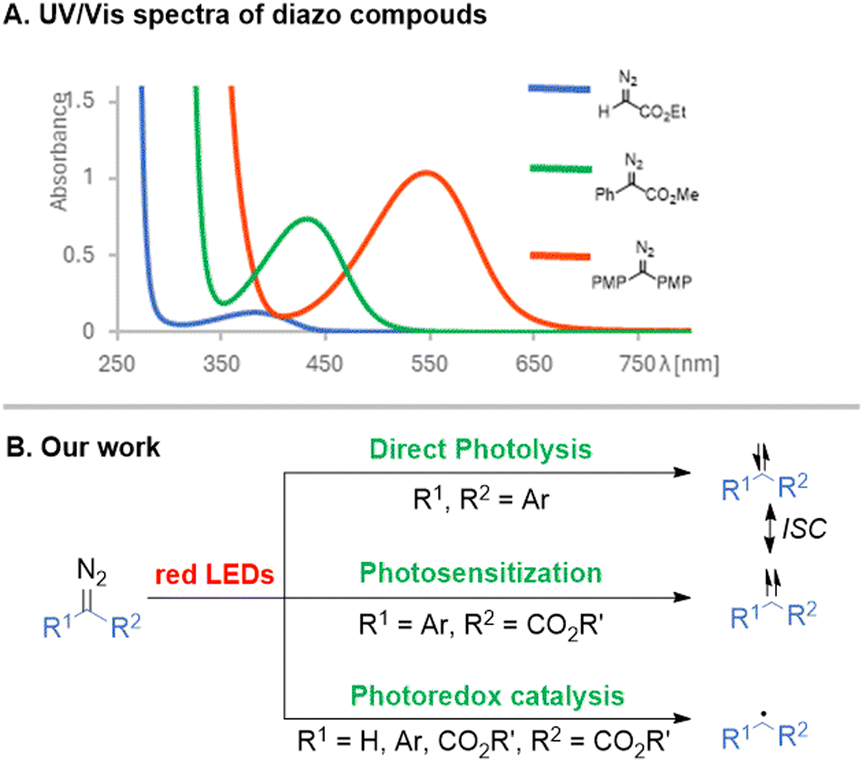 | ||
| Fig. 1 Red light-induced reactions of diazoalkanes. | ||
Photolysis – Direct photolysis of diazoalkanes enables carbene generation with no catalyst required. Although acceptor-only and acceptor/acceptor diazo compounds exhibit light absorption beyond the visible range, replacing H/one of the acceptor groups with an aryl substituent bathochromically shifts the λmax toward the visible spectrum.16,32 By increasing the donating character of the phenyl ring, λmax is shifted even further (for –OMe, λmax = 543 nm),33 and has an impact on the carbene spin state. Given the ubiquity of free hydroxy-, amino-, and thio-groups in natural compounds, we focused on red light-induced photolysis of diaryldiazoalkanes in the presence of alcohols, amines, and thiols (Scheme 1). The light-induced method works well for primary alcohols efficiently affording ethers 1–5. Incorporation into the phenolic O–H bond, a tyrosine model, also proved successful (6, 72%). Secondary and tertiary alcohols were slightly less effective (7, 8 and 10), but cholesterol derivative 9 formed almost quantitatively. On the other hand, irradiation of diaryl diazoalkane bearing amino groups at p-positions (λmax = 566 nm)34 in the presence of benzyl alcohol led to product 11 in a diminished yield (45%). Noteworthily, when beneficial, the substrate ratio could be reversed and diazoalkane excess could be used instead. Benzyl and aromatic amines formed in a slightly diminished yield, in contrast with productive amine 15 formation (84%). 2-Hydroxypyridine gave a mixture of O–H and N–H insertion products but, upon isolation, full conversion to amide 18 occurred. The scope of tolerated thiols is broad, and even thiophenol and bulky adamantanethiol efficiently furnished products 20 and 21. Furthermore, the feasibility of the method was examined with N-Boc protected cysteine ester, and insertion occurred on both the N–H and S–H bonds (23, 74%).
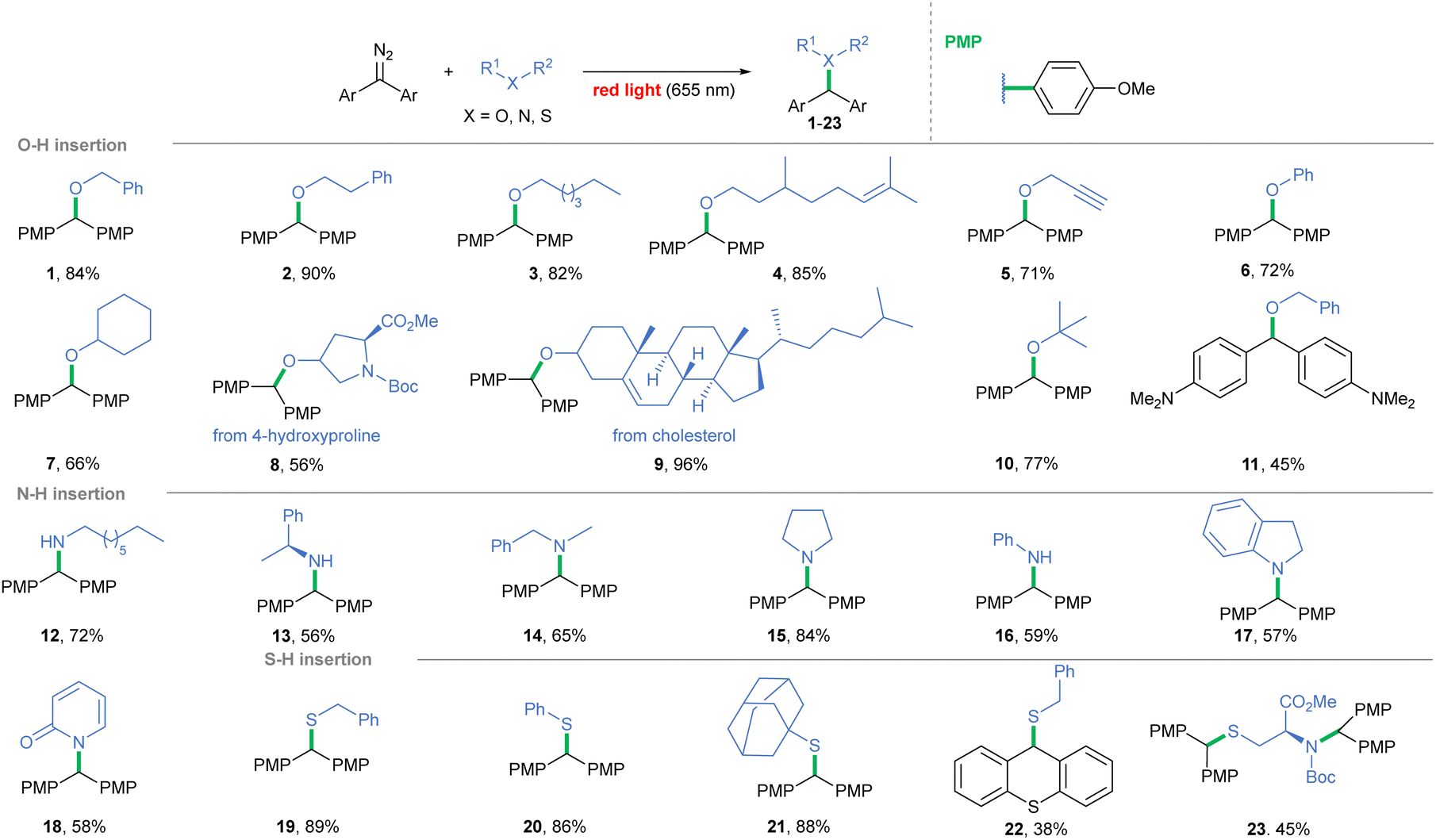 | ||
| Scheme 1 Scope of red light-induced carbene insertion into the X–H bond. | ||
Photosensitization – Most carbene precursors, including diazo compounds, do not, however, absorb red light (Fig. 1A), and for their activation photocatalytic approaches are required. Among these, photosensitization with the use of a dye of proper ET level gives access to triplet excited states via triplet–triplet energy transfer (EnT).35,36 Only recently, mild Ir-sensitized strategies to access triplet carbenes from diazirines and 1,3,4-oksadiazolines under blue light irradiation were proposed by MacMillan and our group,30,37 but approaches relying on red light remain challenging. Porphyrins are sensitizers widely applied in photooxidation, photodynamic therapy and artificial photosynthesis.38–41 We tested these red-light-absorbing organic dyes for photosensitization of diazoalkanes. When aryldiazoesters (ET ≈ 133 kJ mol−1, calculated using SMD(DCM)/M06/6-311++g(d,p)//B3LYP-D3/6-31g(d)), were irradiated with red light in the presence of H2TPP (ET = 138 kJ mol−1)42 and oxygen, β-ketoesters 24–26 formed (Scheme 2A).
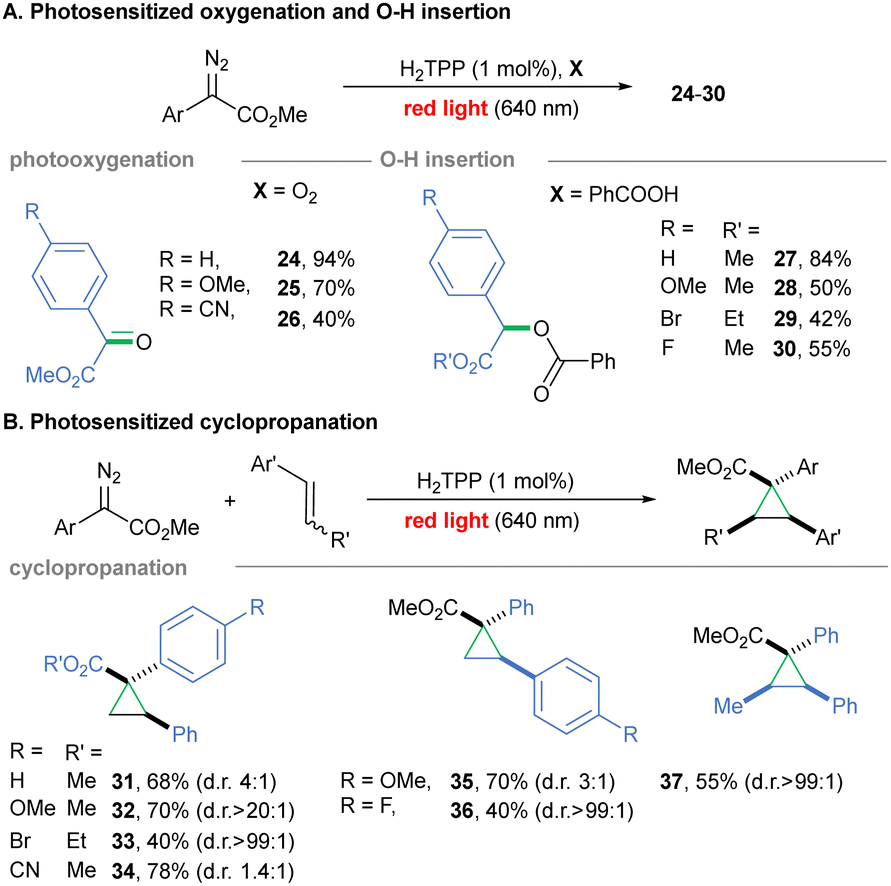 | ||
| Scheme 2 Red light-induced photosensitized transformations of diazo compounds. | ||
For more electrophilic aryl diazoalkane, a loss of selectivity was observed (26, 40%). Since porphyrins are well-known 1O2 sensitizers,38,39 maintaining oxygen-free conditions was crucial to prevent competitive oxidation pathways in consecutive O–H insertion (Scheme 2A) and cyclopropanation (Scheme 2B, see ESI†). The insertion into O–H carboxylic bonds works for various aryldiazoesters leading to products 27–30. Electron-poor aryldiazo ester reached the highest cyclopropanation productivity (34, 90%). The method is suitable for both electron-rich and -poor styrenes, with a better outcome for p-methoxy-styrene-derived product 35 (70%). A modest yield was observed when the internal olefin was subjected to the reaction conditions giving cyclopropane 37 (55%). α-Diazo esters, diazomalonates, and aryldiazoketones possessing higher ET values than porphyrin (calculated ET = 158 kJ mol−1 for EDA) cannot, in principle, be activated under the developed conditions. Intuitively, the reaction rate for diazoalkane transformation depends on the carbene rate formation, which for the red light-mediated EnT-approach occurs slower than via direct photolysis under blue light (see ESI†).
Photoredox catalysis – To unlock the red-light mediated reactivity of yet unconquered α-diazo esters, we screened the possibilities offered by photoredox catalysis. These acceptor-only types of diazoalkanes are reduced to alkyl radicals via proton coupled electron transfer (PCET, ERED = −1.28 V vs. SCE for EDA).43 In this view, numerous blue light-induced methodologies utilizing diazoesters as surrogates of alkyl radicals have been reported.43–48 Recently, we have proved that porphyrins are suitable photo-oxidants and photo-reductants for red light-mediated organic transformations.31 Therefore, we harnessed their photoredox abilities to tune already reported blue light-induced, radical-based transformations of α-diazoesters and applied them on red illumination instead. Our studies were initiated with the redesign of the photocatalyzed synthesis of γ-oximino esters, originally performed by Li under blue light with the use of α-diazoester, styrene and TBN as starting materials.43 Optimization studies substantially shortened the reaction time (reported on blue: 60 h) to 37 h by thermally accelerating the isomerization of the nitroso compound to the final product 38 (see ESI†). Our method works well for various α-diazoesters giving esters 38, 40 and 41 in yields comparable to those reported by Li (Scheme 3A). A slight yield decrease was observed for trans-anethole, though with a similar E/Z ratio (product 39). Due to solubility problems, the synthesis of pregnenolone-derived ester 42 was less efficient. For the Ru-catalyzed reaction a key step relies on the reduction of diazo ester by the photocatalyst in the excited state. In our case, as the reduction potential of the porphyrin in the excited state (–0.91 vs. SCE)31 is higher than that of EDA (–1.28 V vs. SCE), we assume that the excited porphyrin oxidizes DIPEA, thus generating a strongly reducing porphyrin radical anion, similar to the mechanism reported for the generation of radicals from aminopyridinium salts.49
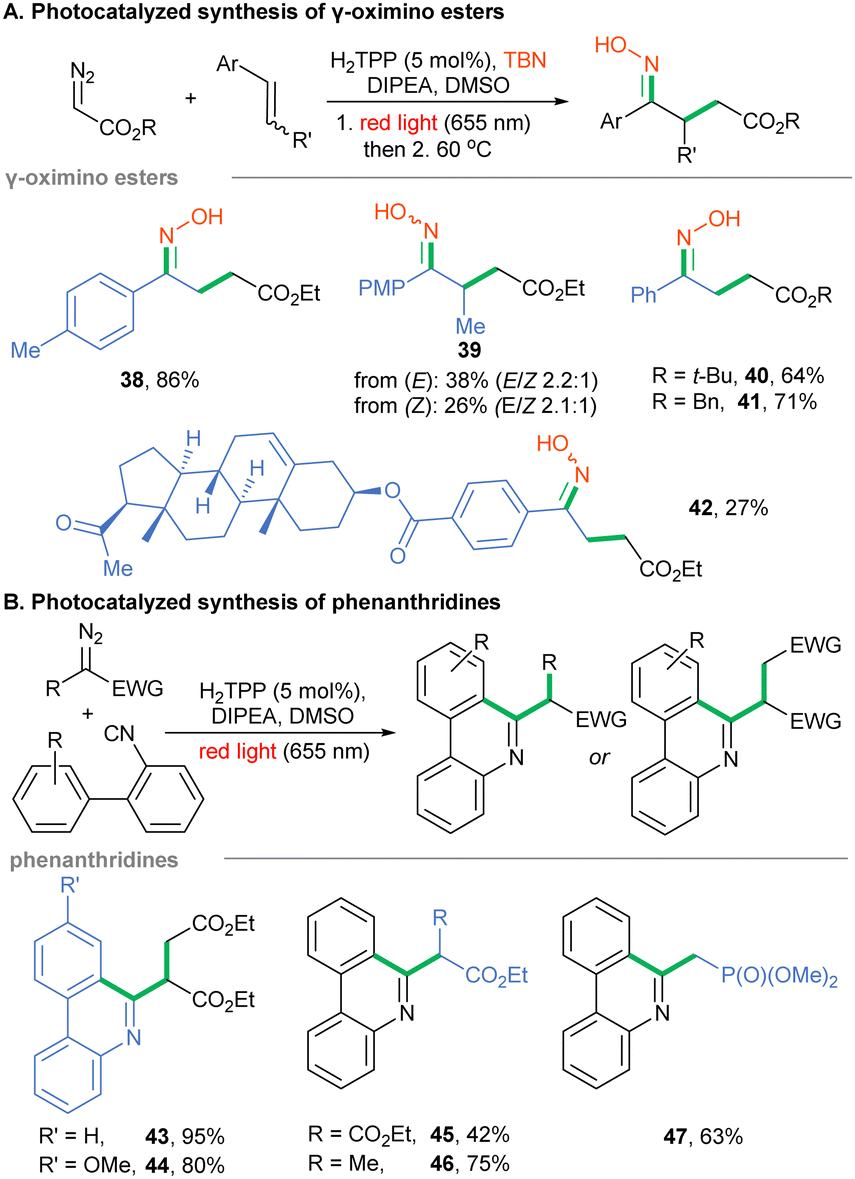 | ||
| Scheme 3 Diazo compounds as radical precursors in red light-mediated photocatalyzed transformations. | ||
Next, we examined an analogous PCET-based approach toward phenanthridines utilizing isocyanobiphenyls and diazoalkanes.50Scheme 3B shows the optimization of the red light-mediated protocol-enabled synthesis of heterocycles 43–47 with better productivity or comparable to the Xuan methodology. Finally, there are methodologies involving diazo reagents in which the diazo moiety remains intact or does not generate reactive intermediates. To fill the picture of the photochemistry of diazo compounds under red-light irradiation, such transformations were studied. Given that sole H2TPP is unable to photoreduce EDA, we tested H2TPP as a photo-oxidant of diversely substituted tetrahydroisoquinolines in the presence of EDA and red light, similar to Zhou's report.51 In fact, products 48–51 were obtained in decent yields (Scheme 4A). Furthermore, diazo compounds have been shown to react with radicals generated under photochemical conditions, including alkyl radicals generated from NHPI esters in the presence of Rose Bengal on yellow LEDs.52 We performed this transformation with the H2TPP catalyst instead, under red light irradiation. A wide range of donor/acceptor diazoalkanes reacted under the developed conditions to give hydrazones 52–56 (Scheme 4B).
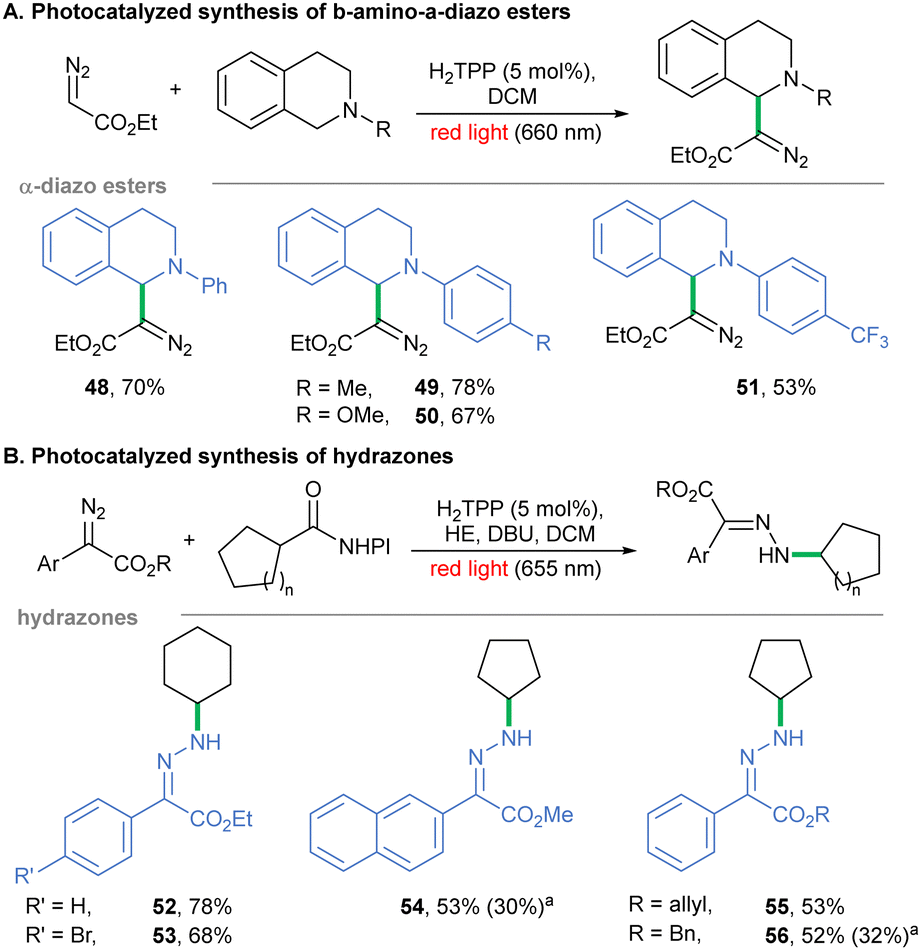 | ||
| Scheme 4 Diazo compounds as radical acceptors in red light-mediated photocatalyzed transformations. aReaction set under blue light irradiation (25 W, 455 nm). | ||
In summary, this study demonstrates that photochemistry provides tools for red light-driven activation of various diazo compounds. A proper structural modification of diazoalkane results in a bathochromic shift of the absorption maxima allowing for direct photolysis under low-energetic, red-light irradiation. If this pathway is not possible, we induce transformations of diazo compounds taking advantage of nature-inspired dyes, established as safe and effective for photodynamic therapy and artificial photosynthesis. The triplet energy level of the porphyrin excited state is sufficient for productive EnT to aryl-diazo esters giving access to triplet carbenes. Other diazoalkanes may be activated through porphyrin-mediated photoredox processes by undergoing reduction to alkyl radicals or by serving as radical acceptors. Therefore, three-modes of activation of diazo compounds under red-light irradiation have been unlocked.
Financial support for this work was provided by National Science Center, Poland, grant number Maestro UMO-2020/38/A/ST4/00185.
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS PubMed.
- C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 651 CrossRef CAS PubMed.
- N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952 CrossRef CAS PubMed.
- S. Jia and E. M. Sletten, ACS Chem. Biol., 2022, 17, 3255 CrossRef CAS PubMed.
- J. Li, H. Kong, C. Zhu and Y. Zhang, Chem. Sci., 2020, 11, 3390 RSC.
- B. D. Fairbanks, L. J. Macdougall, S. Mavila, J. Sinha, B. J. Kirkpatrick, K. S. Anseth and C. N. Bowman, Chem. Rev., 2021, 121, 6915 CrossRef CAS PubMed.
- G. S. Kumar and Q. Lin, Chem. Rev., 2021, 121, 6991 CrossRef CAS PubMed.
- W. Song, Y. Wang, J. Qu, M. M. Madden and Q. Lin, Angew. Chem., Int. Ed., 2008, 47, 2832 CrossRef CAS PubMed.
- W. Song, Y. Wang, J. Qu and Q. Lin, J. Am. Chem. Soc., 2008, 130, 9654 CrossRef CAS PubMed.
- P. An, T. M. Lewandowski, T. G. Erbay, P. Liu and Q. Lin, J. Am. Chem. Soc., 2018, 140, 4860 CrossRef CAS PubMed.
- Z. Yu, T. Y. Ohulchanskyy, P. An, P. N. Prasad and Q. Lin, J. Am. Chem. Soc., 2013, 135, 16766 CrossRef CAS PubMed.
- P. Lederhose, Z. Chen, R. Muller, J. P. Blinco, S. Wu and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2016, 55, 12195 CrossRef CAS PubMed.
- C. Wang, H. Zhang, T. Zhang, X. Zou, H. Wang, J. E. Rosenberger, R. Vannam, W. S. Trout, J. B. Grimm, L. D. Lavis, C. Thorpe, X. Jia, Z. Li and J. M. Fox, J. Am. Chem. Soc., 2021, 143, 10793 CrossRef CAS PubMed.
- N. Sellet, M. Cormier and J. P. Goddard, Org. Chem. Front., 2021, 8, 6783 RSC.
- Z. Yang, M. L. Stivanin, I. D. Jurberg and R. M. Koenigs, Chem. Soc. Rev., 2020, 49, 6833 RSC.
- J. Durka, J. Turkowska and D. Gryko, ACS Sustainable Chem. Eng., 2021, 9, 8895 CrossRef CAS.
- Ł. W. Ciszewski, K. Rybicka-Jasińska and D. Gryko, Org. Biomol. Chem., 2019, 17, 432 RSC.
- H. M. L. Davies and J. R. Denton, Chem. Soc. Rev., 2009, 38, 3061 RSC.
- S.-S. Ge, B. Chen, Y.-Y. Wu, Q.-S. Long, Y.-L. Zhao, P.-Y. Wanga and S. Yang, RSC Adv., 2018, 8, 29428 RSC.
- S. Harada, Chem. Pharm. Bull., 2021, 69, 1170 CrossRef CAS PubMed.
- K. A. Mix, M. R. Aronoff and R. T. Raines, ACS Chem. Biol., 2016, 11, 3233 CrossRef CAS PubMed.
- Z. Liu and F. H. Arnold, Curr. Opin. Biotechnol, 2021, 69, 43 CrossRef CAS PubMed.
- B. J. Wittmann, A. M. Knight, J. L. Hofstra, S. E. Reisman, S. B. J. Kan and F. H. Arnold, ACS Catal., 2020, 10, 7112 CrossRef CAS PubMed.
- R. Mao, D. J. Wackelin, C. S. Jamieson, T. Rogge, S. Gao, A. Das, D. M. Taylor, K. N. Houk and F. H. Arnold, J. Am. Chem. Soc., 2023, 145, 16176 CrossRef CAS PubMed.
- D. C. Miller, R. G. Lal, L. A. Marchetti and F. H. Arnold, J. Am. Chem. Soc., 2022, 144, 4739 CrossRef CAS PubMed.
- K. Chen, X. Huang, S. B. Jennifer Kan, R. K. Zhang and F. H. Arnold, Science, 2018, 360, 71 CrossRef CAS PubMed.
- R. K. Zhang, K. Chen, X. Huang, L. Wohlschlager, H. Renata and F. H. Arnold, Nature, 2019, 565, 67 CrossRef CAS PubMed.
- A. Blencowe and W. Hayes, Soft Matter, 2005, 1, 178 RSC.
- S. W. Huth, J. V. Oakley, C. P. Seath, J. B. Geri, A. D. Trowbridge, D. L. Parker Jr., F. P. Rodriguez-Rivera, A. G. Schwaid, C. Ramil, K. A. Ryu, C. H. White, O. O. Fadeyi, R. C. Oslund and D. W. C. MacMillan, J. Am. Chem. Soc., 2023, 145, 16289 CrossRef CAS PubMed.
- J. B. Geri, J. V. Oakley, T. Reyes-Robles, T. Wang, S. J. McCarver, C. H. White, F. P. Rodriguez-Rivera, D. L. Parker Jr., E. C. Hett, O. O. Fadeyi, R. C. Oslund and D. W. C. MacMillan, Science, 2020, 367, 1091 CrossRef CAS PubMed.
- K. Rybicka-Jasińska, T. Wdowik, K. Łuczak, A. J. Wierzba, O. Drapała and D. Gryko, ACS Org. Inorg. Au, 2022, 2, 422 CrossRef PubMed.
- I. D. Jurberg and H. M. L. Davies, Chem. Sci., 2018, 9, 5112 RSC.
- S. Jana, C. Pei, C. Empel and R. M. Koenigs, Angew. Chem., Int. Ed., 2021, 60, 13271 CrossRef CAS PubMed.
- R. W. R. Humphreys and D. R. Arnold, Can. J. Chem., 1979, 57, 2652 CrossRef CAS.
- F. Strieth-Kalthoff and F. Glorius, Chem, 2020, 6, 1888 CAS.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190 RSC.
- K. Orłowska, J. V. Santiago, P. Krajewski, K. Kisiel, I. Deperasińska, K. Zawada, W. Chaładaj and D. Gryko, ACS Catal., 2023, 13, 1964 CrossRef.
- R. Costa e Silva, L. O. da Silva, A. de Andrade Bartolomeu, T. J. Brocksom and K. T. de Oliveira, Beilstein J. Org. Chem., 2020, 16, 917 CrossRef PubMed.
- J. Kou, D. Dou and L. Yang, Oncotarget, 2017, 8, 81591 CrossRef PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890 CrossRef CAS PubMed.
- R. Seely, Photochem. Photobiol., 1978, 27, 639 CrossRef.
- L. G. Arnaut, Design of porphyrin-based photosensitizers for photodynamic therapy, in Advances in Inorganic Chemistry, ed. R. van Eldik and G. Stochel, Academic Press, 2011, ch. 5, vol. 63, pp. 187–233 Search PubMed.
- Y. Liu, K. Zhu, J. Zhao and P. Li, Org. Lett., 2022, 24, 6834 CrossRef CAS PubMed.
- F. Li, S. Zhu and R. M. Koenigs, Chem. Commun., 2022, 58, 7526 RSC.
- Ł. W. Ciszewski, J. Durka and D. Gryko, Org. Lett., 2019, 21, 7028 CrossRef PubMed.
- X. Huang, R. D. Webster, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 12636 CrossRef CAS PubMed.
- Y.-L. Su, G. X. Liu, L. De Angelis, R. He, A. Al-Sayyed, K. S. Schanze, W.-H. Hu, H. Qiu and M. P. Doyle, ACS Catal., 2022, 12, 1357 CrossRef CAS.
- Y. L. Su, G. X. Liu, J. W. Liu, L. Tram, H. Qiu and M. P. Doyle, J. Am. Chem. Soc., 2020, 142, 13846 CrossRef CAS PubMed.
- K. Goliszewska, K. Rybicka-Jasińska, J. A. Clark, V. I. Vullev and D. Gryko, ACS Catal., 2020, 10, 5920 CrossRef CAS.
- H. B. Ye, X. Y. Zhou, L. Li, X. K. He and J. Xuan, Org. Lett., 2022, 24, 6018 CrossRef CAS PubMed.
- T. Xiao, L. Li, G. Lin, Z. W. Mao and L. Zhou, Org. Lett., 2014, 16, 4232 CrossRef CAS PubMed.
- C. M. Chan, Q. Xing, Y. C. Chow, S. F. Hung and W. Y. Yu, Org. Lett., 2019, 21, 8037 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc05174a |
| This journal is © The Royal Society of Chemistry 2023 |