
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic C(sp3)–H thiolation by a double SH2 strategy using thiosulfonates†
Nobukazu
Taniguchi
 *a,
Mamoru
Hyodo
*b,
Lin-Wei
Pan
c and
Ilhyong
Ryu
*bc
*a,
Mamoru
Hyodo
*b,
Lin-Wei
Pan
c and
Ilhyong
Ryu
*bc
aFaculty of Liberal Arts, Sciences and Global Education, Osaka Metropolitan University, Sakai, Osaka 599-8531, Japan. E-mail: ntaniguchi@omu.ac.jp
bInstitute for Research Promotion, Osaka Metropolitan University, Sakai, Osaka 599-8531, Japan. E-mail: hyodo_mam@omu.ac.jp; ryu@omu.ac.jp
cDepartment of Applied Chemistry, National Yang Ming Chiao Tung University, Hsinchu 30010, Taiwan. E-mail: ryu@nycu.edu.tw
First published on 23rd November 2023
Abstract
Site-selective C(sp3)–H thiolation using thiosulfonates has been achieved using the decatungstate anion as a photocatalyst. Using the protocol, a variety of thiolated compounds were synthesized in good yields. The transformation consists of a cascade of double SH2 reactions, HAT and ArS group transfer, and PCET (proton-coupled electron transfer) of the leaving arylsulfonyl radical to arylsulfinic acid thus allowing the catalyst, W10O324−, to be recovered.
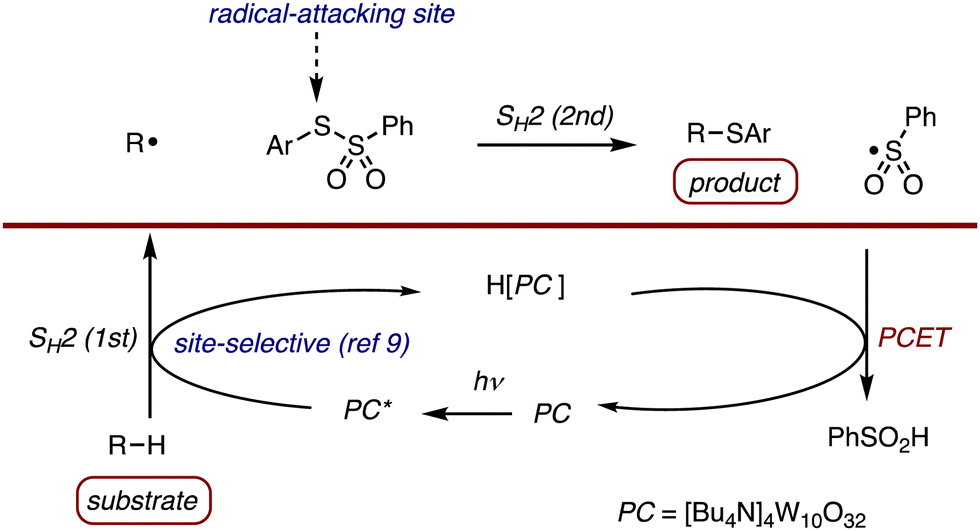
C(sp3)–S bonds are widely present in natural products, pharmaceuticals and value-added compounds.1 Accordingly, the development of synthetic methods for C–S bond formation is highly important in organic synthesis and in chemical biology.2 It is known that thiosulfonates3 act as an excellent unimolecular chain transfer (UMCT) reagent4 delivering an arylthio group to carbon-centred radicals in an SH2 (bimolecular homolytic substitution) manner resulting in the formation of thiolated compounds with liberation of arylsulfonyl radicals (Scheme 1, top equation).5 Despite the broad applicability of radical thiolation reactions that involve the use of thiosulfonates, they have rarely been used for C(sp3)–H thiolation except for a formyl C–H bond and an α-C–H bond of oxygen, nitrogen and a phenyl group.6,7 The use of the photocatalytic8 reactions using decatungstate anion has excellent potential for use in site-selective C(sp3)–H functionalization reaction.9 The origin is the control of the SH2 (HAT: hydrogen atom transfer) transition states based on synergistic polar and steric effects to give the desired alkyl radicals. Combined with the excellent capability of a PhSO2 group to function as an efficient radical leaving group, recent efforts of C–H/C–C conversion include photocatalytic C–H alkenylation,10 alkynylation,11,12 imination,12,13 cyanation,14 allylation12,15 and heteroarylation.16 We hypothesized that a photocatalyzed strategy using a double SH2 concept (HAT plus SH2 at sulfur atom) using phenylthiobenzenesulfonate as a thiolating reagent and decatungstate anion as a catalyst would be promising (Scheme 1) for allowing for site-selective C–H thiolation to be achieved.
 | ||
| Scheme 1 Double SH2 strategy for site-selective C(sp3)–H thiolation using thiosulfonates and photocatalyst. | ||
To establish optimal conditions for this reaction, we initially surveyed a photocatalytic C–H thiolation using cyclohexane 1.1 and PhSSO2Ph 2.1 in the presence of 2 mol% of decatungstate anion as a model reaction (Scheme 2). For a light source we examined a power-adjustable high-power LED (365 nm, 480 W, MiChS-UV-LED-s)17 and a low-power LED (365 nm, 3.4 W) (PENN PhD Photoreactor M2, Sigma-Aldrich). The former gave a 47% yield of the desired phenylthiolated cyclohexane 3.1 after photoirradiation for 1 h (entry 1), while the latter gave 38% after 1 h (entry 4) and 65% after 8 h (entry 6). Photoirradiation without catalyst did not give 3.1 (entry 5). The addition of K2CO3 as a base increased the yield of 3.1 to 78% yield (entry 2). A shorter reaction time of 30 min caused the yield to be decreased to 64% (entry 3). We also tested blacklight irradiation (15 W, 16 h) but was not so effective (entry 6).
 | ||
| Scheme 2 Photocatalytic C–H phenylthiolation of cyclohexane 1.1 using S-phenyl benzenethiosulfonate 2.1. | ||
On the basis of the protocol of entry 2, we then investigated the scope and limitations of the photocatalytic C(sp3)–H thiolation for a wide range of organic compounds, the results for which are summarized in Table 1. The thiolation of cyclic alkanes such as cyclohexane, cyclopentane, cycloheptane, cyclooctane and cyclododecane gave thioethers 3.1 (78%), 3.2 (58%), 3.3 (61%), 3.4 (54%) and 3.5 (27%), respectively. The use of p-chlorotoluene gave the corresponding benzyl thioether 3.6 in 42% yield. The thiolation of linear ethers such as diethyl ether, t-butyl methyl ether, ethyl phenolate, anisole, and p-chloroanisole afforded the corresponding thioethers 3.7 (69%), 3.8 (38%), 3.9 (35%), 3.10 (42%) and 3.11 (36%). In the reaction of p-methylanisole, the ether α-C–H reacted preferentially compared to the α-benzylic C–H, giving 3.12 as a major product (90/10) in a total yield of 41%. Thioanisole was converted into the dithioacetal 3.13 albeit the modest yield. The reaction of cyclic ethers such as THF, tetrahydropyran and 1,4-dioxane proceeded smoothly to give 3.14 (83%), 3.15 (67%) and 3.16 (67%), respectively. Three different arylthio groups were confirmed to be introduced into 1,4-dioxane using ArS-SO2Ph (Ar = p-MeC6H4, p-ClC6H4, p-BrC6H4), giving 3.17 (85%), 3.18 (78%) and 3.19 (64%), respectively. Using a similar photocatalytic protocol, the use of n-BuS–SO2Ph produced the corresponding butyl sulfide 3.20 in 59% yield. The reaction of 1,3-dioxane gave 3.21 (63%) selectively which was derived from H-abstraction at the 4-position rather than the 2-position. The reaction of 2,2-dimethyl-1,3-dioxolane and 1,3-dioxolane with 2.1 gave 3.22 (71%) and 3.23 (44%), respectively. The reaction of dimethoxyethane gave a 59/41 mixture of 3.24 and 3.24′via methylene and methyl C–H bond cleavage in a 72% total yield. The reaction of dimethoxymethane took place selectively at the methyl-C–H bond to give 3.25 as a sole product in 61% yield.
| a General conditions: 1 (2.5 mmol), 2 (0.5 mmol), (Bu4N)4W10O32 (0.01 mmol), K2CO3 (0.5 mmol), acetonitrile (1 mL), photoirradiation using UV-LED (365 nm, 480 W) under N2 atmosphere for 1 h. Isolated yields after silica gel column chromatography are given. The ratios were determined by 1H NMR. b UV-LED (365 nm, 360 W). c 1.5 h. d (Bu4N)4W10O32: 4 mol%. |
|---|

|
We then examined the thiolation of cyclic ketones and lactones. The reaction of cyclohexanone gave a 67/33 mixture of 3.26 and 3.26′ in 39% yield. In 4-methylcyclohexanone, methine C–H thiolation occurred selectively, giving 3.27 (96/4) in 50% yield. The thiolation of 3,3-dimethylcyclohexanone was site-selective at the 5-methylene to give 3.28 in 43% yield. The thiolation of cyclopentanone gave 3.29 as the sole product in 64% yield. The thiolation of γ-butyrolactone proceeded highly selectively at the methylene bond α to the oxygen (94/6), giving 3.30 in 50% yield. The thiolation of γ-valerolactone took place at the methine C–H to give 3.31 as the sole product in 80% yield. The thiolation of N-acetylpiperidine and N-trifluoroacetylpiperidine proceeded site-selectively to give the thiolation products 3.32 (62%) and 3.33 (31%), respectively. The thiolation of N-acetyl morpholine proceeded exclusively at the N-methylene group, giving the corresponding thioether 3.34 in 47% yield. The thiolation of N,N-dimethylacetamide took place selectively at the N-methyl group to give 3.35 in 83% yield. The thiolation of N-methyl-2-pyrrolidinone occurred highly selectively at the α-methylene C–H bond to the α-methyl C–H bond, to give 3.36 as the major product (91/9) in 65% total yield. The thiolation of N,N-dimethylformamide (DMF) gave the methyl-thiolation product 3.37 in 42% yield. This is in contrast with the reaction of DMF with thiosulfonate using Rhodamine B as a photocatalyst, giving thiocarbamate via the generation of a carbamoyl radical.18,19 The scalability of the present protocol was confirmed by the reaction of 1.1 using 5 mmol of 2.1 t, which gave 71% (685 mg) yield of 3.1 (see ESI†).
Concerning the 1,3-dioxolanes, 3.21, 3.23 and 3.25, interestingly, the thiolation products at α-C–H bonds that were activated only by a single oxygen atom were obtained selectively. To avoid a possibility of the degradation of products we used milder irradiation (blacklight, 30 W) for the reaction of 1,3-dioxolane with 2.1 (Scheme 3, eqn (1)). As our expectation, the reaction was sluggish and after 18 h it gave 20% yield of 3.23 as only thiolated product together with a large amount of 2.1 remained unreacted. Using similar photoirradiation conditions, we carried out decatunsgtate-catalyzed alkenylation of 1,3-dioxolane using 4,10 which gave a mixture of alkenylated products 5 and 5′ (Scheme 3, eqn (2)), suggesting the first SH2 takes place at both C–H bonds to generate radical A and B. Accordingly, we concluded that the second SH2 with thiosulfone 2.1 is inefficient for radical B stabilized by two adjacent oxygens, rendering the site-selective C(sp3)–H thiolation to give 3.23.20
 | ||
| Scheme 3 Comparison of C(sp3)–H thiolation and alkenylation for 1,3-dioxolane. | ||
Finally, we examined the flow photocatalytic reaction (Scheme 4),21 using a flow setup consisting of a tubular PFA reactor (inner diameter = 1 mm, outer diameter = 1.56 mm, length = 3.82 m, total inner volume = 3 mL) and high-power LED (365 nm, 480 W, MiChS UV-LED-s). An acetonitrile solution containing 2.1 (500 mM) and the decatungstate anion (2 mol%) was mixed with neat cyclohexane 1.1 using a T-shaped mixer with a 400 μm inner diameter (MiChS α400) and introduced into a photo flow reactor connected with a back pressure regulator (5 psi). Irradiation with a residence time of 30 min resulted in the formation of the envisaged cyclohexyl phenyl sulfide 3.1 in 71% isolated yield.
 | ||
| Scheme 4 Flow photocatalytic thiolation. | ||
In summary, we have reported herein on a photocatalytic protocol for the thiolation of C(sp3)–H bonds using thiosulfonates 2. The decatungstate anion functions as an excellent photocatalyst for introducing sulfur-functions to organic molecules 1. The site-selective thiolation protocol is applicable to a wide range of functionalized aliphatic compounds, such as ethers, ketones, esters, ethers and amides, thus greatly expanding the utility of radical-based C(sp3)–H thiolation chemistry.
We wish to thank JSPS for Grant-in Aid for Scientific Research (B) (no. 19H02722). I. R. thanks funding from the National Science and Technology Council, Taiwan (NSTC 112-2113-MA49-013) and the Centre for Emergent Functional Matter Science at NYCU for support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) N. Wang, P. Saidhareddy and X. Jiang, Nat. Prod. Rep., 2020, 37, 246–275 RSC; (b) C. Bharathi, K. J. Prabahar, C. S. Prasad, R. M. Srinivasa, G. N. Trinadhachary, V. K. Handa, R. Dandala and A. Naidu, Pharmazie, 2008, 63, 14–19 CAS; (c) S. Raghavan, V. Krishnaiah and B. Sridhar, J. Org. Chem., 2010, 75, 498–501 CrossRef CAS PubMed; (d) I. Mohammed, I. R. Kummetha, G. Singh, N. Sharova, G. Lichinchi, J. Dang, M. Stevenson and T. M. Rana, J. Med. Chem., 2016, 59, 7677–7682 CrossRef CAS PubMed; (e) A. Dondoni, Angew. Chem., Int. Ed., 2008, 47, 8995–8997 CrossRef CAS PubMed; (f) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (g) V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J.-M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS PubMed.
- (a) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2022, 122, 16110–16239 CrossRef CAS PubMed; (b) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596–1636 CrossRef CAS PubMed; (c) T. Kondo and T.-a Mitsudo, Chem. Rev., 2000, 100, 3205–3220 CrossRef CAS PubMed; (d) C.-F. Lee, Y.-C. Liu and S. S. Badsara, Chem. – Asian J., 2014, 9, 706–722 CrossRef CAS PubMed; (e) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400–5449 CrossRef CAS PubMed.
- For a recent review of ArSSO2Ar, see: P. Mampuys, C. R. McElroy, J. H. Clark, R. V. A. Orru and B. U. W. Maes, Adv. Synth. Catal., 2020, 362, 3–64 CrossRef CAS.
- (a) D. P. Curran, J. Xu and E. Lazzarini, J. Am. Chem. Soc., 1995, 117, 6603–6604 CrossRef CAS; (b) D. P. Curran, J. Xu and E. Lazzarini, J. Chem. Soc., Perkin Trans. 1, 1995, 3049–3059 RSC.
- (a) H. Li, C. Shan, C.-H. Tung and Z. Xu, Chem. Sci., 2017, 8, 2610–2615 RSC; (b) Y. Dong, P. Ji, Y. Zhang, C. Wang, X. Meng and W. Wang, Org. Lett., 2020, 22, 9562–9567 CrossRef CAS PubMed; (c) W. Li and L. Zhou, Org. Lett., 2022, 24, 3976–3981 CrossRef CAS PubMed; (d) X. Zhou, Z. Peng, P. G. Wang, Q. Liu and T. Jia, Org. Lett., 2021, 23, 1054–1059 CrossRef CAS PubMed; (e) T. Song, H. Li, F. Wei, C.-H. Tung and Z. Xu, Tetrahedron Lett., 2019, 60, 916–919 CrossRef CAS; (f) Z. Peng, H. Yin, H. Zhang and T. Jia, Org. Lett., 2020, 22, 5885–5889 CrossRef CAS PubMed; (g) Y. Liu, N. Zhang, Y. Xu and Y. Chen, J. Org. Chem., 2021, 86, 16883–16891 Search PubMed; (h) M. R. Mutra, G. V. S. Kudale, J. Li, W.-H. Tsai and J.-J. Wang, Green Chem., 2020, 22, 2288–2300 RSC.
- (a) W.-Z. Bi, W.-J. Zhang, C.-Y. Li, L.-H. Shao, Q.-P. Liu, S.-X. Feng, Y. Geng, X.-L. Chen and L.-B. Qu, Org. Biomol. Chem., 2022, 20, 3902–3906 RSC; (b) Z. Ye, Z. Lei, X. Ye, L. Zhou, Y. Wang, Z. Yuan, F. Gao and R. Britton, J. Org. Chem., 2022, 87, 765–775 CrossRef CAS PubMed; (c) J. Yan, H. Tang, E. J. R. Kuek, X. Shi, C. Liu, M. Zhang, J. L. Piper, S. Duan and J. Wu, Nat. Commun., 2021, 12, 7214 CrossRef CAS PubMed; (d) W.-Z. Bi, W.-J. Zhang, Z.-J. Li, Y.-H. He, S.-X. Feng, Y. Geng, X.-L. Chen and L.-B. Qu, Org. Biomol. Chem., 2021, 19, 8701–8705 RSC; (e) E. André-Joyaux, A. Kuzovlev, N. D. C. Tappin and P. Renaud, Angew. Chem., Int. Ed., 2020, 59, 13859–13864 CrossRef PubMed; (f) Y. Zhang, P. Ji, W. Hu, Y. Wei, H. Huang and W. Wang, Chem. – Eur. J., 2019, 25, 8225–8228 CrossRef CAS PubMed; (g) Also see a review: X.-M. Xu, D.-M. Chen and Z.-L. Wang, Chin. Chem. Lett., 2020, 31, 49–57 CrossRef CAS.
- Direct thiolation of unactivated C–H bonds is rare, see: (a) Y. Li, F. Zhu, Z. Wang and X.-F. Wu, Chem. – Asian J., 2016, 11, 3503–3507 CrossRef CAS PubMed; (b) R. Mao, S. Bera, A. C. Turla and X. Hu, J. Am. Chem. Soc., 2021, 143, 14667–14675 CrossRef CAS PubMed . Also see C–H sulfinylation: ; (c) H. Tan, C. Zhang, Y. Deng, M. Zhang, X. Cheng and J. Wu, Org. Lett., 2023, 25, 2883–2888 CrossRef CAS PubMed.
- For selected recent works on decatungstate catalysis, see: (a) P. J. Sarver, N. B. Bissonnette and D. W. C. MacMillan, J. Am. Chem. Soc., 2021, 143, 9737–9743 CrossRef CAS PubMed; (b) F. Babawale, K. Murugesan, R. Narobe and B. König, Org. Lett., 2022, 24, 4793–4797 CrossRef CAS PubMed; (c) Z. Ye, Z. Lei, X. Ye, L. Zhou, Y. Wang, Z. Yuan, F. Gao and R. Britton, J. Org. Chem., 2022, 87, 765–775 CrossRef CAS PubMed.
- For a perspective on site-selective C–H functionalization by decatungstate catalysis, see: D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa and I. Ryu, ACS Catal., 2018, 8, 701–713 CrossRef CAS.
- Y.-T. Wang, Y.-L. Shih, Y.-K. Wu and I. Ryu, Adv. Synth. Catal., 2022, 364, 1039–1043 CrossRef CAS.
- L. Capaldo and D. Ravelli, Org. Lett., 2021, 23, 2243–2247 CrossRef CAS PubMed.
- Y.-L. Shih, S.-H. Huang, Y.-K. Wu and I. Ryu, Bull. Soc. Chem. Jpn, 2022, 95, 1501–1505 CrossRef CAS.
- X. Wang, M. Yu, H. Song, X. Liu and Q. Wang, Org. Lett., 2021, 23, 8353–8358 CrossRef CAS PubMed.
- F. K. Kim, S. Lee and S. H. Hong, Org. Lett., 2021, 23, 5501–5508 CrossRef PubMed.
- L. Qiao, X. Fu, Y. Si, X. Chen, L. Qu and B. Yu, Green Chem., 2022, 24, 5614–5619 RSC.
- (a) J. Xu, L. Liu, Z.-C. Yan, Y. Liu, L. Qin, N. Deng and H.-J. Xu, Green Chem., 2023, 25, 2268–2273 RSC . Also see azidation: ; (b) Y.-C. Lu, S.-C. Kao and J. G. West, Chem. Commun., 2022, 58, 4869–4872 RSC.
- M. Hyodo, H. Iwano, T. Kasakado., T. Fukuyama and I. Ryu, Micromachines, 2021, 12, 1307–1315 CrossRef PubMed.
- W.-Z. Bi, W.-J. Zhang, Z.-J. Li, Y.-H. He, S.-X. Feng, Y. Geng, X.-L. Chen and L.-B. Qu, Org. Biomol. Chem., 2021, 19, 8701–8705 RSC.
- The decatunsgtate-photocatalytic reaction of DMF with alkenes results in site-selective addition of the N-methyl C–H bond, see: (a) S. Angioni, D. Ravelli, D. Emma, D. Dondi, M. Fagnoni and A. Albini, Adv. Synth. Catal., 2008, 350, 2209–2214 CrossRef CAS; (b) M. C. Quattrini, S. Fujii, K. Yamada, T. Fukuyama, D. Ravelli, M. Fagnoni and I. Ryu, Chem. Commun., 2017, 53, 2335–2338 RSC.
- 2-(Phenylthio)-1,3-dioxolane is thermally stable, see: C. S. Shiner, T. Tsunoda, B. A. Goodman, S. Ingham, S.-H. Lee and P. E. Vorndam, J. Am. Chem. Soc., 1989, 111, 1381–1392 CrossRef CAS.
- For flow decatungstate photocatalysis, see: (a) F. Bonassi, D. Ravelli, S. Protti and M. Fagnoni, Adv. Synth. Catal., 2015, 357, 3687–3695 CrossRef CAS; (b) D. M. Schultz, F. Levesque, D. A. DiRocco, M. Reibarkh, Y. Ji, L. A. Joyce, J. F. Dropinski, H. Sheng, B. D. Sherry and I. W. Davies, Angew. Chem., Int. Ed., 2017, 56, 15274–15278 CrossRef CAS PubMed; (c) G. Laudadio, Y. Deng, K. van del Wal, D. Ravelli, M. Nuno, M. Fagnoni, D. Guthrie, Y. Sun and T. Noel, Science, 2020, 369, 92–96 CrossRef CAS PubMed; (d) S. Bonciolini, M. D. Filippo and M. Baumann, Org. Biomol. Chem., 2020, 18, 9428–9432 RSC; (e) M. Hyodo, H. Iwano, T. Kasakado, T. Fukuyama and I. Ryu, Micromachines, 2021, 12, 1307–1315 CrossRef PubMed; (f) Y.-L. Shi, Y.-K. Wu, M. Hyodo and I. Ryu, J. Org. Chem., 2023, 88, 6548–6552 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data. See DOI: https://doi.org/10.1039/d3cc05149h |
| This journal is © The Royal Society of Chemistry 2023 |