
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of core–shell polymer particles in supercritical carbon dioxide via iterative monomer addition†
Kristoffer
Kortsen‡
 a,
Morgan
Reynolds-Green‡
a,
Bradley
Hopkins
ab,
Alison
McLellan
a,
Matthew J.
Derry
c,
Paul D.
Topham
c,
Jeremy J.
Titman
a,
Daniel J.
Keddie
a,
Vincenzo
Taresco
a and
Steven M.
Howdle
*a
a,
Morgan
Reynolds-Green‡
a,
Bradley
Hopkins
ab,
Alison
McLellan
a,
Matthew J.
Derry
c,
Paul D.
Topham
c,
Jeremy J.
Titman
a,
Daniel J.
Keddie
a,
Vincenzo
Taresco
a and
Steven M.
Howdle
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: steve.howdle@nottingham.ac.uk
bDepartment of Chemical and Environmental Engineering, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
cAston Advanced Materials Research Centre, Aston University, Aston Triangle, Birmingham, B4 7ET, UK
First published on 14th November 2023
Abstract
A new, robust methodology for the synthesis of polystyrene–poly(methyl methacrylate) (PS–PMMA) core–shell particles using seeded dispersion polymerisation in supercritical carbon dioxide is reported, where the core–shell ratio can be controlled predictably via manipulation of reagent stoichiometry. The key development is the application of an iterative addition of the MMA shell monomer to the pre-prepared PS core. Analysis of the materials with differing core–shell ratios indicates that all are isolated as single particle populations with distinct and controllable core–shell morphologies.
Polymer particles with core–shell morphologies have a wide range of applications such as surface coatings, printing, catalysis, impact modifiers, pollution control, sensing and drug delivery.1–4 The interest in core–shell particles derives from their superior properties which arise from the combined characteristics of the core and shell materials.5
Core–shell particles are made up of a simple two-component system, consisting of a gas, liquid or solid core encased within a solid shell, typically with a diameter in the nano or micro size range.5–9 For the synthesis of polymer-based core–shell particles a broad range of monomers have been successfully employed, including (meth)acrylates, styrenes and acrylamides.5,10
The synthesis of polymeric core–shell particles in conventional solvents is typically achieved via waterborne dispersed phase polymerization processes, such as emulsion, dispersion and precipitation polymerization methods.5 While these syntheses can be tailored to provide a range of morphologies, isolation of pure polymers by removal of the aqueous continuous phase is energy intensive and can lead to wastewater contamination.11–13
Supercritical carbon dioxide (scCO2) is an alternative to standard solvents, possessing some unique properties; above its critical point (31.1 °C, 73.8 bar) carbon dioxide exhibits a lack of surface tension, enhanced diffusivity, and adjustable density. ScCO2 is also non-toxic and non-flammable. In addition, most vinylic monomers are soluble in scCO2, whereas their polymers are not. This eliminates the need for energy-intensive drying or creation of solvent wastes from precipitation during purification after polymerization; the monomer is simply removed with the scCO2 upon depressurisation.14 Indeed, these properties of scCO2 have been exploited for the synthesis of polymer materials,15–20 their impregnation with a range of species,15,16,21 and the extraction of additives from their internal structure.22
As mentioned above, synthesis of core–shell particles in conventional solvents, such as water, has been widely explored. Significant scope however remains to explore their synthesis in scCO2. There are some prior reports in this area, but to date development of a scalable method that delivers control over the shell thickness relative to the core has not been achieved. For example, Cao et al. synthesised poly(NIPAM) microgel particles in scCO2 using complex stabiliser molecules that functioned as a stabilising shell.6,23 While they did produce microgel particles with temperature and pH responsiveness, the shell was in fact the stabiliser from the emulsion system; the overall layer thickness was not demonstrated and true core–shell morphology was not achieved. In a second example, our group previously prepared particles bearing a poly(benzyl acrylate) (PBzA) shell deposited on a poly(methyl methacrylate) (PMMA) core which was successfully synthesised for BzA loadings of 27 wt% with respect to MMA monomer in scCO2. While this initial sample possessed a core–shell morphology, increases in BzA loading resulted in a change of particle morphology rather than increased shell thickness; small internal domains of PBzA were observed surrounded by a continuous PMMA phase. This was attributed to the supercritical fluid's ability to plasticise and swell PMMA, allowing the increasingly more abundant PBzA monomer to permeate into the intended core phase.24
Herein, we report a new method, based on seeded dispersion polymerisation, to prepare polymer-based particles with tailored core–shell morphologies, using PS and PMMA as exemplar polymers, where the core–shell ratio can easily be varied by simple manipulation of the reaction conditions, as illustrated by the preparation of particles with core–shell mass ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:2, 1:4, and 1:8. Key to this new methodology is iterative addition of the MMA monomer to the preprepared PS core. This ensures polymerisation occurs on the preformed PS-core particles,§25,26 preventing secondary nucleation and the formation of unwanted PMMA particle byproducts.
:1, 1:2, 1:4, and 1:8. Key to this new methodology is iterative addition of the MMA monomer to the preprepared PS core. This ensures polymerisation occurs on the preformed PS-core particles,§25,26 preventing secondary nucleation and the formation of unwanted PMMA particle byproducts.
Brief details of the syntheses are provided below (see ESI† for further details). Firstly, the PS core was synthesised via radical polymerisation (RP), using AIBN initiator and PDMS-MA stabiliser in scCO2 at 65 °C and 245 bar for 24 hours (Fig. 1(a)). The synthesised polystyrene was re-dispersed in scCO2 at 65 °C and 245 bar for 15 hours, after which stock solutions of AIBN in MMA were injected into the PS suspension. Successive AIBN-MMA injections (i.e. iterative monomer addition) were carried out at 4 hour intervals (Fig. 1(b)). It is necessary to allow the complete polymerisation and hence consumption of the previously injected AIBN-MMA prior to carrying out subsequent injections; excess MMA with respect to preformed particles favours the formation of additional discrete PMMA particles (see Fig. S1, ESI†).
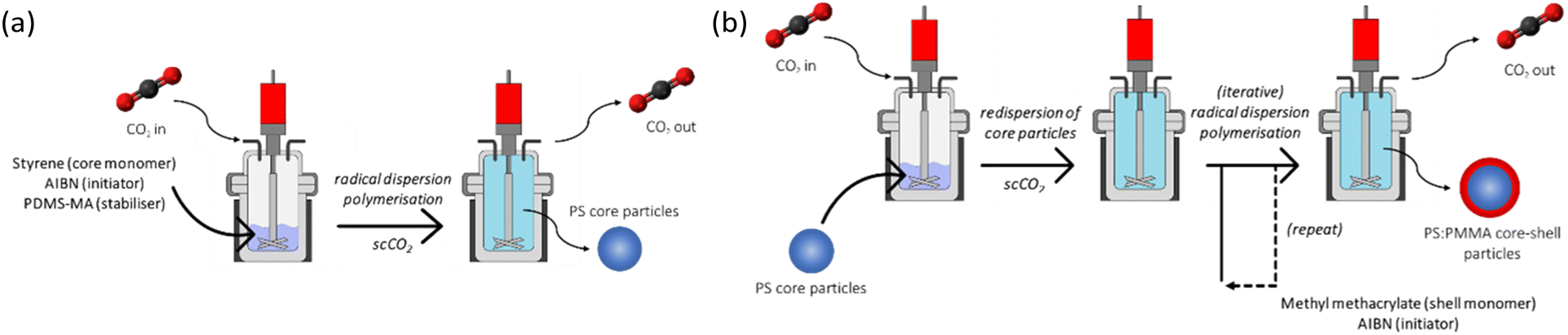 | ||
| Fig. 1 Reaction schematic of (a) PS core and (b) PMMA shell syntheses in supercritical CO2 in a high-pressure autoclave. | ||
1H NMR (see Fig. S2, ESI†) determined that monomer conversion during PMMA shell synthesis was high (>93%) in all cases (see Table 1). Additionally, 1H–13C CP-MAS NMR analysis of dried particles demonstrated an increase in PMMA-shell derived signals relative to PS-core signals, consistent with the targeted mass ratios (see Fig. 2(a)).
| Entry | PS:PMMA mass ratio |
MMA conv.b (%) | M n (kDa) | Đ | d SEM (nm) | σ d (nm) | d DLS (nm) | PDIe | d calc. (nm) | T g,DSC (°C) | T g,DMA (°C) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Targeted | Experimentala | PS | PMMA | PS | PMMA | |||||||||
| a Calculated from 1H NMR and polymer densities (ρPS = 1.05; ρPMMA = 1.19).27 b Calculated from 1H NMR. c From GPC analysis (THF eluent, PMMA standards). d Average diameter (dSEM) and standard deviation of diameter (σd) of particles from SEM analysis. e Average diameter (dDLS) and polydispersity index (PDI) of particles from DLS. f Calculated theoretical diameter obtained from a predictive model (see ESI for more information). g T g transition not observed due to low relative content of PS. | ||||||||||||||
| 1 | PS core | — | — | 17 | 2.9 | 373 | 77 | 356 | 0.146 | 256 | 99 | — | 103 | — |
| 2 | 1:1 |
1:1.0 |
93 | 36 | 2.4 | 470 | 136 | 401 | 0.307 | 448 | 93 | 120 | 108 | 124 |
| 3 | 1:2 |
1:1.9 |
98 | 40 | 2.8 | 509 | 179 | 486 | 0.303 | 513 | 99 | 122 | 108 | 120 |
| 4 | 1:4 |
1:3.7 |
98 | 44 | 2.1 | 641 | 160 | 552 | 0.361 | 608 | —g | 120 | 108 | 128 |
| 5 | 1:8 |
1:8.4 |
99 | 50 | 2.0 | 736 | 173 | 740 | 0.750 | 740 | —g | 120 | 108 | 128 |
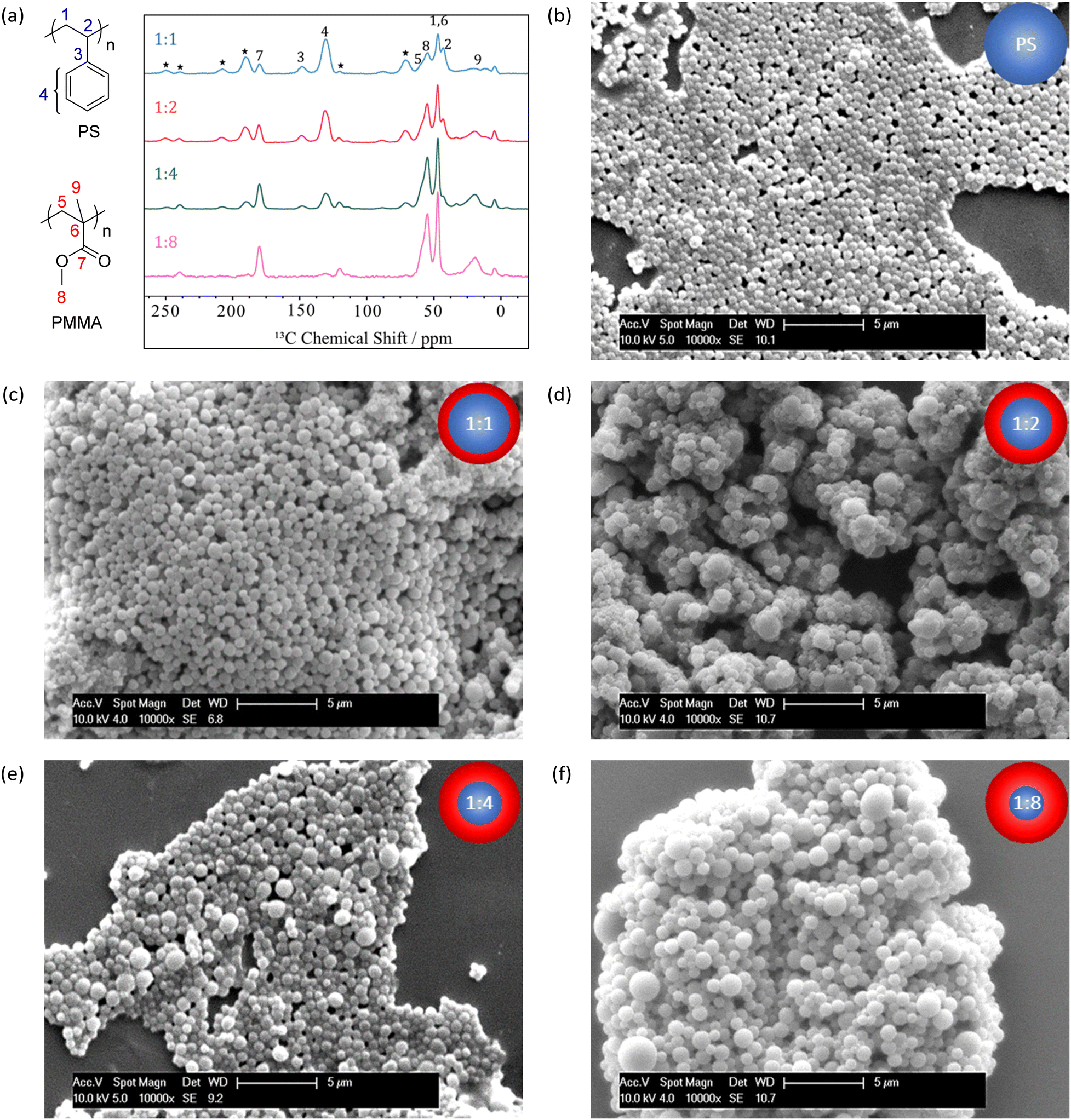 | ||
| Fig. 2 (a) 1H–13C CP-MAS spectra of PS–PMMA core–shell polymers with PS:PMMA ratios of 1:1, 1:2, 1:4 and 1:8 (note: spinning sidebands marked with *, inset shows numbering of 13C assignments), and SEM micrographs of (b) PS particles (core), and PS–PMMA core–shell particles with targeted mass ratios of (c) 1:1, (d) 1:2, (e) 1:4 and (f) 1:8. | ||
Scanning electron microscopy (SEM) and dynamic light scattering (DLS) illustrated that relatively uniform particles that were larger than the initial PS core particles had been produced, and that the diameter predictably increased with an increasing MMA monomer feed (see Table 1 and Fig. 2(b)–(f)). A simple predictive model was devised to calculate the diameter of the various core–shell particles, based on assumptions of perfect sphere growth and a consistent density throughout both phases of the particles (see Scheme S1, ESI†). Close agreement between this predictive model and experimentally obtained diameters by SEM and DLS further confirm formation of the desired core–shell morphology (see Table 1 and Fig. S3, ESI†). Additional corroboration of this morphology by small angle X-ray scattering (SAXS) was attempted at diamond light source,28 however there was insufficient scattering length density contrast to discern the shell thickness (see Fig. S4, ESI†).
Thermal properties of the core–shell particles were analysed by differential scanning calorimetry (DSC) and dynamic mechanical analysis (DMA). DSC indicated that both PS and PMMA were present in the final product, as their respective glass transition temperatures (Tgs) could be observed, ∼95 °C for PS and ∼125 °C for PMMA (see Fig. S5, ESI†). DMA analyses illustrated a dual transition in the stiffness of the core–shell particles, indicating both polymers were present in different phases (see Fig. 3(a)). To verify this dual transition was the result of the desired core–shell morphology, the materials were compared to physical, unprocessed blends of PS and PMMA particles, prepared at the same mass ratios.¶ Indeed, DMA traces indicated the thermal properties of core–shell particles differ from those of the physical blends. The Tgs of components within the core–shell systems were found to converge, presumably due to molecular interactions at the interface. As expected, the blends displayed discrete Tgs of the individual components (see Fig. S6, ESI†).
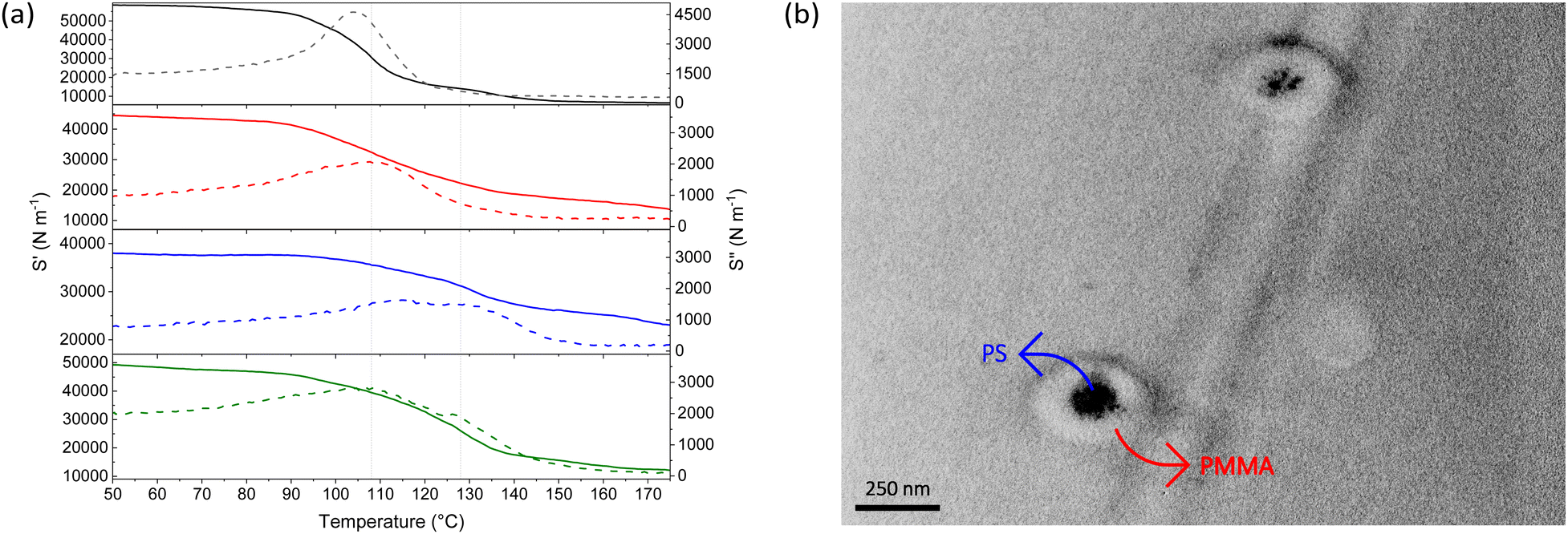 | ||
| Fig. 3 (a) Storage stiffness (S′) (solid lines) and loss stiffness (S′′) (dashed lines) for core–shell particles with PS:PMMA targeted mass ratios of 1:1 (black lines), 1:2 (red lines), 1:4 (blue lines) and 1:8 (green lines) as function of temperature (note: the guide lines at the Tgs of PS (108 °C) and PMMA (124 °C)), and (b) labelled TEM micrograph of the PS–PMMA core–shell particles with targeted mass ratios of 1:1 (note: the PS core has been selectively stained with RuO4 vapour to show the core–shell morphology). | ||
To obtain further evidence of core–shell morphology, the particles were viewed by transmission electron microscopy (TEM). The particles were stained with ruthenium tetroxide (RuO4)29 which provides clear contrast between the PS core and PMMA shell, due to interaction of RuO4 with the aromatic rings of PS (see Fig. 3(b)).
This communication reports the first successful synthesis of core–shell particles in supercritical carbon dioxide, where the core–shell ratio can be controlled predictably. Key to the success of this new methodology is the iterative addition of the MMA during the preparation of the shell to the pre-prepared PS core particles. SEM and DLS demonstrate that there is a single particle population indicating PS and PMMA are co-located within the same particles, whilst DSC, DMA and TEM confirm that two distinct polymer phases were present within the particles. This new method for preparation of tailored core- shell materials opens up a route to the preparation of bespoke particles that could be used for a broad range of applications. Importantly, the exploitation of scCO2 as a solvent delivers a greener process than conventional methods, eliminating some chemical hazards and minimizing the energy requirements for product isolation and purification.
We would like to thank the EPSRC for their support (EPSRC Centre for Doctoral Training in Sustainable Chemistry; EP/L015633/1). V. T. would like to thank the University of Nottingham for his Nottingham Research Fellowship. The authors would also like to thank the Nanoscale and Microscale Research Centre (nmRC) at the University of Nottingham for access to electron microscope instruments and sample preparation equipment. We would also like to thank Mr Richard Wilson and Mr Mark Guyler for their technical support with the pressure equipment. This work was carried out with the support of Diamond Light Source, instrument I22 (proposal sm33098), and we would like to acknowledge the support received by Dr Andy Smith and all I22 beamline staff during our experiment.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. D. Jones and L. A. Lyon, Macromolecules, 2003, 36, 1988–1993 CrossRef CAS.
- J. S. Downey, R. S. Frank, W.-H. Li and H. D. H. Stöver, Macromolecules, 1999, 32, 2838–2844 CrossRef CAS.
- N. Sasa and T. Yamaoka, Adv. Mater., 1994, 6, 417–421 CrossRef CAS.
- A. K. Khan, B. C. Ray and S. K. Dolui, Prog. Org. Coat., 2008, 62, 65–70 CrossRef CAS.
- R. A. Ramli, W. A. Laftah and S. Hashim, RSC Adv., 2013, 3, 15543–15565 RSC.
- L.-Q. Cao, L.-P. Chen, P.-Y. Cui and J.-D. Wang, J. Appl. Polym. Sci., 2008, 108, 3843–3850 CrossRef CAS.
- X. Fu, L. Bao, J. Lei and J. Wang, J. Vinyl Addit. Technol., 2016, 22, 452–459 CrossRef CAS.
- J.-S. Lee and F.-C. Chang, Polym. Eng. Sci., 2004, 44, 1885–1889 CrossRef CAS.
- W. Schärtl, Adv. Mater., 2000, 12, 1899–1908 CrossRef.
- B. R. Saunders and B. Vincent, Adv. Colloid Interface Sci., 1999, 80, 1–25 CrossRef CAS.
- S. Kirsch, A. Doerk, E. Bartsch, H. Sillescu, K. Landfester, H. W. Spiess and W. Maechtle, Macromolecules, 1999, 32, 4508–4518 CrossRef CAS.
- J. Yang, Y. Mu and X. Li, Mater. Lett., 2022, 322, 132493 CrossRef CAS.
- L. Y. Wang, Y.-J. Lin and W.-Y. Chiu, Synth. Met., 2001, 119, 155–156 CrossRef CAS.
- J. L. Kendall, D. A. Canelas, J. L. Young and J. M. DeSimone, Chem. Rev., 1999, 99, 543–564 CrossRef CAS.
- R. R. Larder, E. Krumins, P. L. Jacob, K. Kortsen, R. Cavanagh, L. Jiang, C. Vuotto, I. Francolini, C. Tuck, V. Taresco and S. M. Howdle, Polym. Chem., 2022, 13, 3768–3779 RSC.
- T. M. Bennett, G. He, R. R. Larder, M. G. Fischer, G. A. Rance, M. W. Fay, A. K. Pearce, C. D. J. Parmenter, U. Steiner and S. M. Howdle, Nano Lett., 2018, 18, 7560–7569 CrossRef CAS.
- M. Alauhdin, T. M. Bennett, G. He, S. P. Bassett, G. Portale, W. Bras, D. Hermida-Merino and S. M. Howdle, Polym. Chem., 2019, 10, 860–871 RSC.
- J. Jennings, M. Beija, A. P. Richez, S. D. Cooper, P. E. Mignot, K. J. Thurecht, K. S. Jack and S. M. Howdle, J. Am. Chem. Soc., 2012, 134, 4772–4781 CrossRef CAS.
- G. He, T. M. Bennett, M. Alauhdin, M. W. Fay, X. Liu, S. T. Schwab, C.-G. Sun and S. M. Howdle, Polym. Chem., 2018, 9, 3808–3819 RSC.
- J. Jennings, G. He, S. M. Howdle and P. B. Zetterlund, Chem. Soc. Rev., 2016, 45, 5055–5084 RSC.
- D. Varga, S. Alkin, P. Gluschitz, B. Péter-Szabó, E. Székely and T. Gamse, J. Supercrit. Fluids, 2016, 116, 111–116 CrossRef CAS.
- R. M. N. Guerra, M. A. L. Marín, A. Sánchez and A. Jiménez, J. Supercrit. Fluids, 2002, 22, 111–118 CrossRef CAS.
- L. Cao, L. Chen, X. Chen, L. Zuo and Z. Li, Polymer, 2006, 47, 4588–4595 CrossRef CAS.
- A. J. Haddleton, T. M. Bennett, X. Chen, R. L. Atkinson, V. Taresco and S. M. Howdle, Polym. Chem., 2020, 11, 5029–5039 RSC.
- P. A. Mueller, G. Storti and M. Morbidelli, Chem. Eng. Sci., 2005, 60, 1911–1925 CrossRef CAS.
- K. Kortsen, H. R. Fowler, P. L. Jacob, E. Krumins, J. C. Lentz, M. R. A. Souhil, V. Taresco and S. M. Howdle, Eur. Polym. J., 2022, 168, 111108 CrossRef CAS.
- Polymer Handbook, ed. J. Brandrup, E. H. Immergut, E. A. Grulke, A. Abe and D. R. Bloch, John Wiley & Sons, 2005 Search PubMed.
- A. J. Smith, S. G. Alcock, L. S. Davidson, J. H. Emmins, J. C. Hiller Bardsley, P. Holloway, M. Malfois, A. R. Marshall, C. L. Pizzey, S. E. Rogers, O. Shebanova, T. Snow, J. P. Sutter, E. P. Williams and N. J. Terrill, J. Synchrotron Rad., 2021, 28, 939–947 CrossRef CAS.
- W. S. J. Li, V. Ladmiral, H. Takeshima, K. Satoh, M. Kamigaito, M. Semsarilar, C. Negrell, P. Lacroix-Desmazes and S. Caillol, Polym. Chem., 2019, 10, 3116–3126 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods and additional characterisation data. See DOI: https://doi.org/10.1039/d3cc04969h |
| ‡ These authors contributed equally. |
| § Due to the low solubility of PMMA in scCO2, polymerisation of MMA is known to (predominantly) occur in the particle (dispersed) phase. See ref. 25 and 26 for more details. |
| ¶ The PMMA particles used in the physical blends were prepared analogously to the PS particles in scCO2 (see ESI†). |
| This journal is © The Royal Society of Chemistry 2023 |