
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ring-expansion and desulfurisation of thiophenes with an aluminium(I) reagent†
Jacob S.
McMullen
,
Andrew J. P.
White
and
Mark R.
Crimmin
 *
*
Department of Chemistry, Imperial College London, 82 Wood Lane, Shepherds Bush, London, W12 0BZ, UK. E-mail: m.crimmin@imperial.ac.uk
First published on 16th November 2023
Abstract
Reactions of thiophene, 2-methylthiophene, 2-methoxythiophene, 2,3-dimethylthiophene, and benzothiophene with the aluminium(I) complex [{ArNC(Me)2H}Al] (Ar = 2,6-di-isopropylphenyl) are reported. In all cases, carbon–sulfur bond activation and ring-expansion of the heterocycle is observed. For thiophene, we identify a reaction network for desulfurisation that includes an unusual second carbon–sulfur bond activation step.
Aromatic heterocycles such as thiophene have been identified as some of the most pervasive sulfur-containing contaminants in petroleum. Their removal is typically achieved through hydrodesulfurisation using heterogeneous catalysts.1 The potential mechanisms underpinning the desulfurisation process are fundamentally interesting. For example, in the case of thiophene, dearomatisation of the heterocycle and breaking of two carbon–sulfur bonds is required to separate the sulfur content from the hydrocarbon chain.
Beginning in the 1960s, there has been keen interest in studying the desulfurisation of thiophenes, and related heterocycles, using homogeneous metal complexes. In early work, it was demonstrated that the complete desulfurisation of thiophene could be achieved by reaction with [Fe3(CO)10].2–4 Subsequent studies provided evidence for a stepwise process that is initiated by insertion of a reactive Fe fragment into the carbon–sulfur bond.5 Numerous examples of this type of reactivity have been reported since and transition metal complexes based on Ti,6 Mo,7,8 Mn9 Fe,10 Ru,11,12 Co,13 Rh,14–20 Ir,21–26 and Pt27–30 are all capable of activating carbon–sulfur bonds of thiophenes. Typically, these reactions are proposed to occur through oxidative addition mechanisms. In cases they can be reversible and C–H bond activation is potentially competitive. That said, the ring-expanded products are usually stable with respect to subsequent carbon–sulfur bond activation step and extrusion of the sulfur atom. There is very limited precedent in which these two discrete events can be observed in a single reaction sequence.
We recently reported that the reaction of 1 with furans led to carbon–oxygen bond activation and ring-expansion (Fig. 1). The selectivity of this reaction could be controlled through addition of a palladium-catalyst and the scope expanded to include dihydrofurans and dihydropyrans.31 Under the same catalytic conditions, thiophene failed to undergo dearomatisation and ring-opening with 1, instead metalation at the 2-position was observed (Fig. 1).32
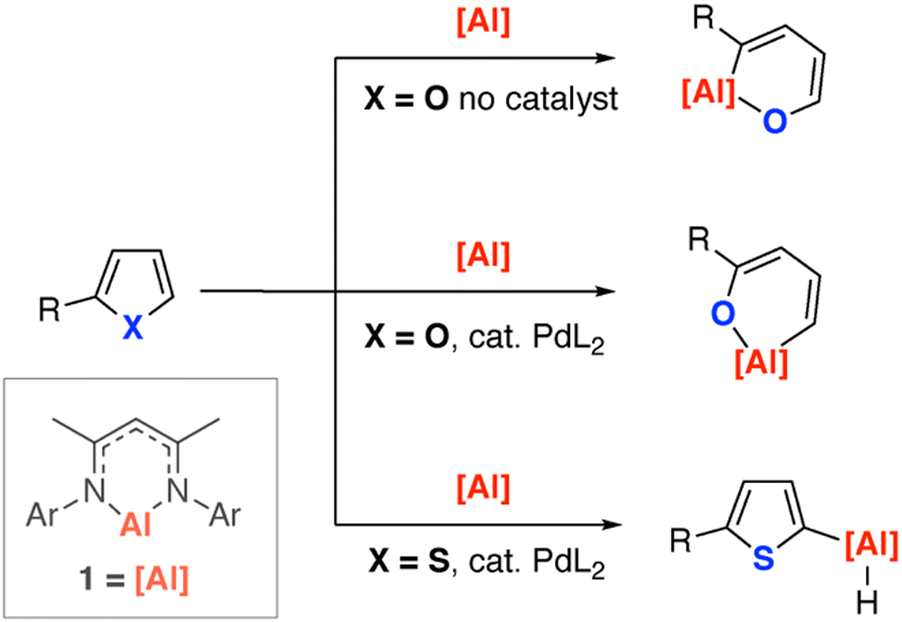 | ||
| Fig. 1 Reactions of an aluminium(I) reagent with furans and thiophenes. Ar = 2,6-di-iso-propylphenyl, L = PCy3. | ||
Here we show that thermal reactions with 1, while complicated, lead to carbon–sulfur bond activation and ring-opening of substituted thiophenes. More importantly, for thiophene itself we identify a reaction network for desulfurisation that evolves from the ring-opened intermediate and includes a unique product derived from a second carbon–sulfur bond activation. Our findings shed light on potential mechanisms in desulfurisation chemistry, and demonstrate, for the first time, well-defined steps in which the two carbon–sulfur bonds of thiophene are broken in sequence.
Reaction of 1 with 1.1 equiv. of thiophene in C6D6 at 60 °C led to a mixture of hydrocarbon soluble products as identified by 1H NMR spectroscopy which formed alongside a colourless crystalline precipitate (Scheme 1). Through variation of the reaction time, the stoichiometry and development of work-up procedures to partially separate mixtures, 2a, 3 and 4 were identified as the hydrocarbon soluble products of this reaction. These results contrast the reaction of 1 with thiophene in the presence of catalytic [Pd(PCy3)2] where C–H activation of the 2-position was observed as a major pathway.32 The insoluble side product was confirmed as the aluminium disulfide complex 5 through isolation and comparison of unit cell data obtained by X-ray diffraction to that in the literature.33
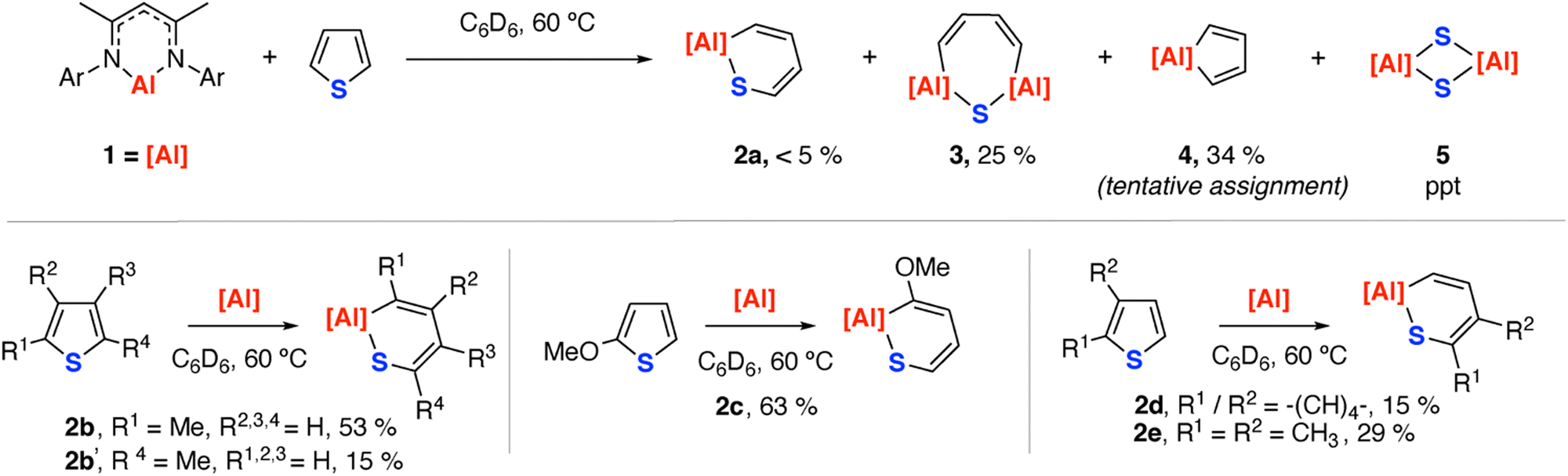 | ||
| Scheme 1 Reactions of thiophenes with aluminium(I) complex [{ArNC(Me)2H}Al] (Ar = 2,6-di-isopropylphenyl) 1. Yields determining by 1H NMR spectroscopy against hexamethyldisiloxane as an internal standard. | ||
Compound 2a is characterised by resonance in the 1H NMR spectrum at δ = 4.87 (s, 1H) corresponding to the β-diketiminate backbone methine. Analogues of 2a can be obtained on reaction of 1 with substituted thiophenes (vide infra).
Compound 3 is unique. Its structure was unambiguously confirmed from a fractional crystallisation and manual separation from the components in the mixture (Fig. 2). In the solid state, 3 exhibits a ternary spirocyclic array with a central 7-membered ring-system that incorporates two aluminium, one sulfur and four carbon atoms. This ring system is derived from the double C–S activation of thiophene by two equiv. of 1. The 7-membered ring system is non-planar and puckers into a distorted twist-boat structure. The Al–S bond lengths of 2.2054(18) and 2.2147(17) Å are consistent with known aluminium(III) sulfide derivatives containing the β-diketiminate ligand system, while Al–C bond lengths of 1.957(5) and 1.977(6) Å are similar to related 5-membered aluminocycles derived from 1.34 Within the hydrocarbon unit, C–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds alternate, consistent with a localised butadiene structure. In C6D6 solution, 3 demonstrates 1H resonances at δ = 6.14 (d, 2H, J = 16.9 Hz) and 7.00 (d, 2H, J = 16.9 Hz) ppm for the butadiene motif. The data are consistent with the retention of a symmetric structure in solution and chemical and magnetic equivalence of sites on opposite sides of the ring.
C bonds alternate, consistent with a localised butadiene structure. In C6D6 solution, 3 demonstrates 1H resonances at δ = 6.14 (d, 2H, J = 16.9 Hz) and 7.00 (d, 2H, J = 16.9 Hz) ppm for the butadiene motif. The data are consistent with the retention of a symmetric structure in solution and chemical and magnetic equivalence of sites on opposite sides of the ring.
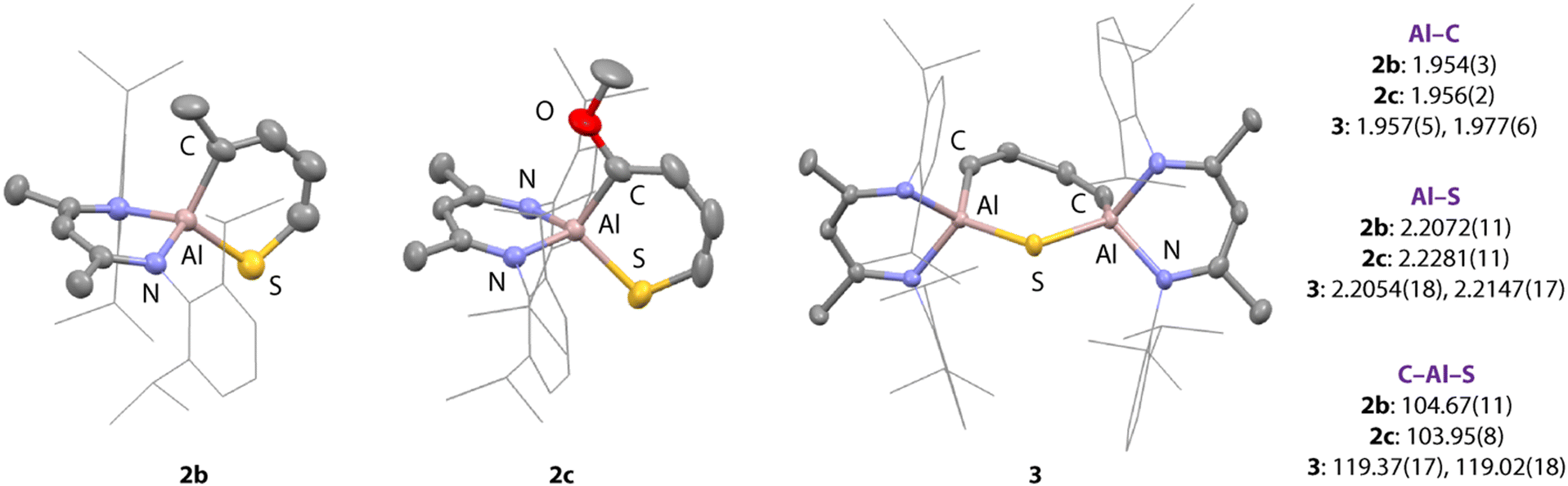 | ||
| Fig. 2 Crystal Structures of 2b, 2c, and 3 Selected bond lengths (Å) and angles (°). | ||
Of the mixture formed on reaction of 1 with thiophene, 4 was the most challenging to unambiguously characterise. This compound could not be isolated, and assignment is tentative. In situ NMR data shows a chemical shift at δ 6.00 (d, 2H, J = 13.7 Hz) ppm, observed in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with the β-diketiminate backbone methine environment at δ 4.90 (s, 1H) ppm, consistent with the proposed formulation. DOSY NMR studies on mixtures containing 3 and 4 are consistent with 4 showing the fastest diffusion coefficient and smallest hydrodynamic radii (D(3) = 8.03 × 10−10 m2 s−1, D(4) = 1.03 × 10−9 m2 s−1), which argue against higher aggregation states or more complex ring-systems. While the formation of 4 is logical based on generation of the desulfurisation product 5, its assignment remains tentative.
:1 with the β-diketiminate backbone methine environment at δ 4.90 (s, 1H) ppm, consistent with the proposed formulation. DOSY NMR studies on mixtures containing 3 and 4 are consistent with 4 showing the fastest diffusion coefficient and smallest hydrodynamic radii (D(3) = 8.03 × 10−10 m2 s−1, D(4) = 1.03 × 10−9 m2 s−1), which argue against higher aggregation states or more complex ring-systems. While the formation of 4 is logical based on generation of the desulfurisation product 5, its assignment remains tentative.
Reactions of 1 with substituted thiophenes were more selective, and in all cases lead to analogues of 2a as the major component in solution (Scheme 1, Fig. 2). For example, 2-methylthiophene forms a 3.5:1 mixture of 2b:2b′ both derived from insertion of the aluminium(I) fragment into a single C–S bond of the ring, with preferred selectivity for the most substituted position. 2-Methoxythiophene reacts similarly, but forms 2c exclusively. This selectivity parallels that found for reactions of 1 with furans. An anomeric effect was proposed as a selectivity influencing factor, with electron-donating groups in the 2-position of the heterocycle acting to weaken the adjacent bond through population of the C–O σ*-orbital. It is likely a similar effect is operating for thiophenes. 1 reacts with benzothiophene to form 2d and 2,3-dimethylthiophene to form 2e. In the latter case, there is no evidence for formation of products from insertion into the most hindered position of the ring, and it is likely the 2,3-disubstitution pattern sterically disfavours the electronically preferred reaction site. Desulfurisation was observed to only occur for thiophene and 2-methylthiophene, with crystals of 5 not being observed during reactions with the other substrates.
The formation of an array of ring-opened and desulfisation products from the reaction of 1 with thiophene raises questions as to their mechanism of formation. A series of DFT calculations were undertaken to better understand these results.‡ The proposed reaction network that leads to the formation of 2a, 3, 4 and 5 is presented in Fig. 3. Formation of 2a is proposed to occur directly from 1 and thiophene (step I). From 2a, either a second C–S bond activation to form 3 (step IIa) or direct desulfurisation to generate 4 and 5 (step IIb) is plausible. 3 is also a potential intermediate for desulfurisation and could lead to the formation of 4 and 5 (step IIIa).
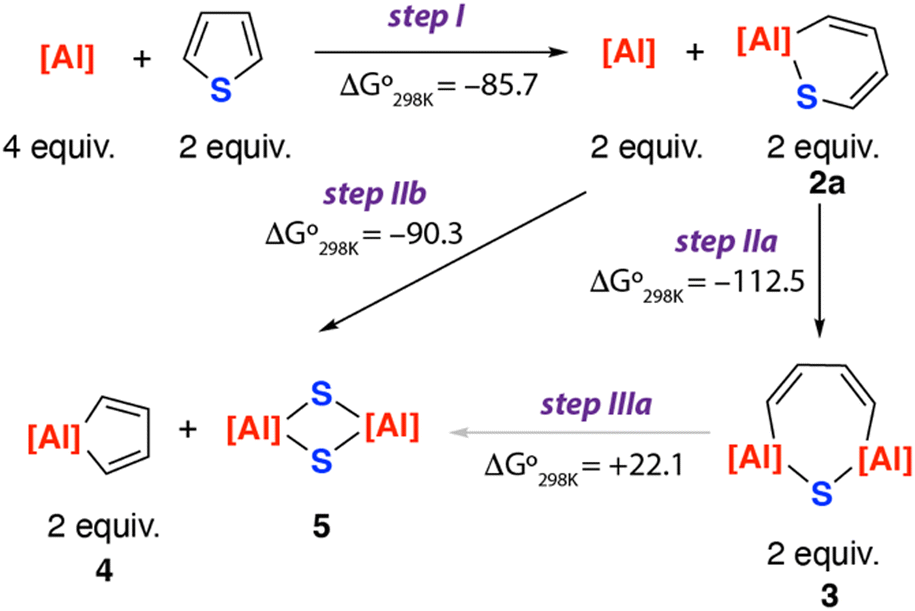 | ||
| Fig. 3 Calculated thermochemistry of the reaction of 1 with thiophene. Values in kcal mol−1. | ||
Consideration of the thermochemistry for each of the reactions, reveals that while steps I, IIa and IIb are all exergonic, step IIIa is considerably endergonic. This suggests that once formed there is little driving force for 3 to react further and it would be expected to be a thermodynamic sink of the reaction sequence. As such it remains more likely that the pathway bifurcates at 2a and this is a common intermediate to form the double C–S activation product 3 and desulfurisation products 4 and 5.
The mechanistic sequence from 1 → 2a → 3 was considered in more detail (Fig. 4). Formation of 2a from 1 and thiophene was calculated to occur through an initial (4+1) cycloaddition (TS-1) to form the [2.1.1] bicycle Int-2 which then undergoes a concerted framework rearrangement (TS-2) with C–S bond activation. The pathway parallels that previously calculated for furan. From 2a, the reaction sequence can repeat. The aluminocycle of 2a contains a butadiene motif which can react again with 1 by a (4+1) cycloaddition (TS-3) to generate a [2.2.1] bicycle Int-4 containing two aluminium sites.
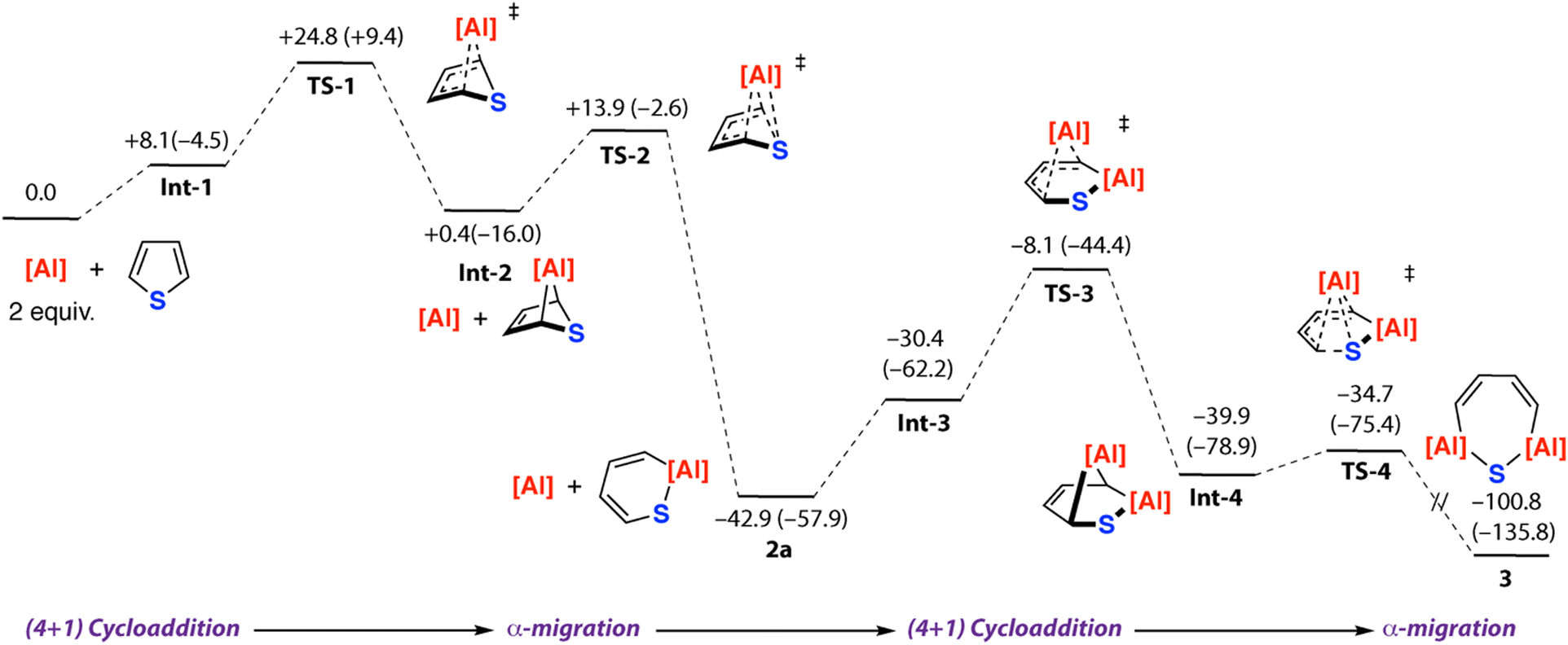 | ||
| Fig. 4 Calculated reaction mechanism for 1st and 2nd C–S activation steps of thiophene with 1. Gibbs free energies, values in kcal mol−1. Enthalpies are given in parentheses in kcal mol−1. | ||
Concerted rearrangement breaks the second C–S bond (TS-4) and opens the bicycle to form 3. The highest barriers on the pathway are those associated with TS-1 and TS-3 which are ΔG‡298K = 24.8 and ΔG‡298K = 34.8 kcal mol−1 respectively. The latter barrier is likely an overestimate of the true experimental barrier and comparison of ΔG‡298K and ΔH‡ values revels there is a significant entropy contribution to this step. Nevertheless, calculations are consistent with the lack of spectroscopic observation of bicyclic intermediates. Sulfur extrusion from 2a could potentially occur from Int-4 from a retro-cycloaddition that forms 4 and a monomeric terminal sulfide, which then dimerises to form 5. While we have not calculated this pathway, related terminal sulfide complexes have recently been reported from reaction of aluminium(I) complexes with sulfur-containing molecules.35
The observation of two sequential C–S bond activation events in the reaction of 1 with thiophene is unique. The result prompted us to re-investigate the addition of 1 to furan, in no case, however, were double C–O activation products observed. While this divergent reactivity is yet to be fully understood, it remains likely that the second insertion transition state TS-3, which brings together two bulky aluminium fragments, is susceptible to steric effects that may be more pronounced in furan derivatives which possess shortened C–X and Al–X bonds compared to the sulfur analogues (X = O, S). Bond strengths may also play an important role with stronger C–O bond in the intermediate being less susceptible to onward reaction that the corresponding species with a C–S bond. In summary, we report the reaction of a low-valent aluminium(I) complex with a series of thiophenes. In all cases, a C–S bond activation and ring-expansion is observed, with products obtain from insertion of the aluminium fragment into the ring system. For thiophene, the reaction does not stop at the 1st C–S activation and a 2nd C–S bond activation and desulfurisation are observed. We are thankful to Imperial College London for support in the form of a President's PhD Scholarship to JSM.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. J. Angelici, Acc. Chem. Res., 1988, 21, 387–394 CrossRef CAS.
- H. D. Kaesz, R. B. King, T. A. Manuel, L. D. Nichols and F. G. A. Stone, J. Am. Chem. Soc., 1960, 82, 4749–4750 CrossRef CAS.
- R. B. King, P. M. Treichel and F. G. A. Stone, J. Am. Chem. Soc., 1961, 83, 3600–3604 CrossRef CAS.
- P. Hübener and E. Weiss, J. Organomet. Chem., 1977, 129, 105–115 CrossRef.
- A. E. Ogilvy, M. Draganjac, T. B. Rauchfuss and S. R. Wilson, Organometallics, 1988, 7, 1171–1177 CrossRef CAS.
- A. Gómez-Torres, J. R. Aguilar-Calderón, C. Saucedo, A. Jordan, A. Metta-Magaña, B. Pinter and S. Fortier, Chem. Commun., 2019, 56, 1545–1548 RSC.
- W. D. Jones, R. M. Chin, T. W. Crane and D. M. Baruch, Organometallics, 1994, 13, 4448–4452 CrossRef CAS.
- D. G. Churchill, B. M. Bridgewater, G. Zhu, K. Pang and G. Parkin, Polyhedron, 2006, 25, 499–512 CrossRef CAS.
- C. A. Dullaghan, S. Sun, G. B. Carpenter, B. Weldon and D. A. Sweigart, Angew. Chem., Int. Ed. Engl., 1996, 35, 212–214 CrossRef CAS.
- I. E. Buys, L. D. Field, T. W. Hambley and A. E. D. McQueen, J. Chem. Soc., Chem. Commun., 1994, 557–558 RSC.
- K. M. K. Dailey, T. B. Rauchfuss, A. L. Rheingold and G. P. A. Yap, J. Am. Chem. Soc., 1995, 117, 6396–6397 CrossRef CAS.
- H. Kawano, H. Narimatsu, D. Yamamoto, K. Tanaka, K. Hiraki and M. Onishi, Organometallics, 2002, 21, 5526–5530 CrossRef CAS.
- W. D. Jones and R. M. Chin, Organometallics, 1992, 11, 2698–2700 CrossRef CAS.
- W. D. Jones and L. Dong, J. Am. Chem. Soc., 1991, 113, 559–564 CrossRef CAS.
- S. Luo, A. E. Skaugset, T. B. Rauchfuss and S. R. Wilson, J. Am. Chem. Soc., 1992, 114, 1732–1735 CrossRef CAS.
- L. Dong, S. B. Duckett, K. F. Ohman and W. D. Jones, J. Am. Chem. Soc., 1992, 114, 151–160 CrossRef CAS.
- C. Bianchini, V. Herrera, M. V. Jimenez, A. Meli, R. Sanchez-Delgado and F. Vizza, J. Am. Chem. Soc., 1995, 117, 8567–8575 CrossRef CAS.
- A. W. Myers, W. D. Jones and S. M. McClements, J. Am. Chem. Soc., 1995, 117, 11704–11709 CrossRef CAS.
- C. Bianchini, P. Frediani, V. Herrera, M. V. Jimenez, A. Meli, L. Rincon, R. Sanchez-Delgado and F. Vizza, J. Am. Chem. Soc., 1995, 117, 4333–4346 CrossRef CAS.
- A. W. Myers and W. D. Jones, Organometallics, 1996, 15, 2905–2917 CrossRef CAS.
- J. Chen, L. M. Daniels and R. J. Angelici, J. Am. Chem. Soc., 1990, 112, 199–204 CrossRef CAS.
- H. E. Selnau and J. S. Merola, Organometallics, 1993, 12, 1583–1591 CrossRef CAS.
- C. Bianchini, A. Meli, M. Peruzzini, F. Vizza, P. Frediani, V. Herrera and R. A. Sanchez-Delgado, J. Am. Chem. Soc., 1993, 115, 2731–2742 CrossRef CAS.
- C. Bianchini, A. Meli, M. Peruzzini, F. Vizza, P. Frediani, V. Herrera and R. A. Sanchez-Delgado, J. Am. Chem. Soc., 1993, 115, 7505–7506 CrossRef CAS.
- C. Bianchini, A. Meli, M. Peruzzini, F. Vizza, S. Moneti, V. Herrera and R. A. Sanchez-Delgado, J. Am. Chem. Soc., 1994, 116, 4370–4381 CrossRef CAS.
- C. Bianchini, J. A. Casares, D. Masi, A. Meli, W. Pohl and F. Vizza, J. Organomet. Chem., 1997, 541, 143–155 CrossRef CAS.
- J. J. Garcia and P. M. Maitlis, J. Am. Chem. Soc., 1993, 115, 12200–12201 CrossRef CAS.
- J. J. Garcia, B. E. Mann, H. Adams, N. A. Bailey and P. M. Maitlis, J. Am. Chem. Soc., 1995, 117, 2179–2186 CrossRef CAS.
- A. Iretskii, H. Adams, P. M. Maitlis, J. J. Garcia and G. Picazo, Chem. Commun., 1998, 61–62 RSC.
- C. A. Dullaghan, X. Zhang, D. L. Greene, G. B. Carpenter, D. A. Sweigart, C. Camiletti and E. Rajaseelan, Organometallics, 1998, 17, 3316–3322 CrossRef CAS.
- T. N. Hooper, R. K. Brown, F. Rekhroukh, M. Garçon, A. J. P. White, P. J. Costa and M. R. Crimmin, Chem. Sci., 2020, 11, 7850–7857 RSC.
- L. Zhang, S. Kaukver, J. McMullen, A. J. P. White and M. R. Crimmin, Organometallics, 2023, 42, 1711–1716 CrossRef CAS.
- V. Jancik, M. M. M. Cabrera, H. W. Roesky, R. Herbst-Irmer, D. Neculai, A. M. Neculai, M. Noltemeyer and H. Schmidt, Eur. J. Inorg. Chem., 2004, 3508–3512 CrossRef CAS.
- C. Bakewell, M. Garçon, R. Y. Kong, L. O’Hare, A. J. P. White and M. R. Crimmin, Inorg. Chem., 2020, 59, 4608–4616 CrossRef CAS PubMed.
- T. Chu, S. F. Vyboishchikov, B. Gabidullin and G. I. Nikonov, Angew. Chem., Int. Ed., 2016, 55, 13306 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation data, and computational details (PDF). X-ray crystallographic data (CIF) and coordinates of stationary points (XYZ). CCDC 2293034–2293036 and 2294820. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc04594c |
| ‡ Details are included in the ESI.† |
| This journal is © The Royal Society of Chemistry 2023 |