
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular intermediates in thermo-mechanochemical direct amidations†
Tomislav
Stolar‡
 *a,
Jasna
Alić
a,
Gregor
Talajić
b,
Nikola
Cindro
b,
Mirta
Rubčić
b,
Krešimir
Molčanov
a,
Krunoslav
Užarević
*a and
José G.
Hernández
*c
*a,
Jasna
Alić
a,
Gregor
Talajić
b,
Nikola
Cindro
b,
Mirta
Rubčić
b,
Krešimir
Molčanov
a,
Krunoslav
Užarević
*a and
José G.
Hernández
*c
aRuđer Bošković Institute, Bijenička c. 54, Zagreb 10000, Croatia. E-mail: krunoslav.uzarevic@irb.hr; tomislav.stolar@gmail.com
bDepartment of Chemistry, Faculty of Science, University of Zagreb, Horvatovac 102a, Zagreb 10000, Croatia
cGrupo Ciencia de los Materiales, Instituto de Química, Facultad de Ciencias Exactas y Naturales, Universidad de Antioquia, Medellín 050010, Colombia. E-mail: joseg.hernandez@udea.edu.co
First published on 20th October 2023
Abstract
We present a solvent-free thermo-mechanochemical approach for the direct coupling of carboxylic acids and amines, which avoids activators and additives. Detailed analysis of the reactions by ex situ and in situ monitoring methods led to the observation, isolation, and characterisation of multicomponent crystalline intermediates that precede the formation of amides. We applied our methodology for the quantitative synthesis of the active pharmaceutical ingredient moclobemide.
The formation of the amide bond is of utmost importance in chemical synthesis. Amide-based compounds find their application in pharmaceuticals,1 agrochemicals,2 polymers,3etc. Notably, around a quarter of marketed drugs contain an amide bond,1 and amide coupling is the most prevalent chemical transformation in drug discovery.4 At the same time, academic and industrial researchers called for improvement in the sustainability of chemical processes that lead to amides as a top priority,5 in line with the principles of green chemistry and sustainable development.6 Among different strategies for the preparation of amides, the most straightforward is the amidation of carboxylic acids and amines (Fig. 1). Their condensation using coupling reagents is the go-to approach for the large-scale preparation of amides.7 However, such an approach suffers from considerable sustainability drawbacks. In the activation step, stoichiometric amounts of coupling reagents are used to activate the carboxylic acids by making the carbonyl group more susceptible to a nucleophilic attack from the amine molecules. Although highly chemically efficient, this approach yields significant amounts of waste and a poor atom economy. Alternatively, solution-based direct catalysed amidation protocols using boron reagents8–10 and group (IV) metal catalysts11–14 are more favourable from the perspective of green chemistry. Yet, catalysed amidations still face a few challenges, such as problems with catalyst loadings, recyclability, and limited substrate scopes. Also, such reactions are usually performed in solution, contributing to waste production.15
 | ||
| Fig. 1 Overview of the main strategies for the amidation of carboxylic acids and amines. | ||
One promising avenue for the atom-efficient synthesis of amides is solvent-free direct thermal amidation. Over 140 years ago, Hoffman reported that amides formed by heating ammonium salts of aliphatic acids at 230 °C and under high pressure.16 Nevertheless, direct thermal amidation has been reported in the literature only scarcely.17 In 1989, Cossy and Pale-Grosdemange reported the synthesis of amides in high yields by heating carboxylic acids and primary amines at 140–180 °C, in the presence of molecular sieves to trap the forming water molecules.18 Jursic and Zdravkovski showed that a variety of amides were formed by heating primary amines and carboxylic acids at 160–180 °C, together with the elimination of water from the reaction mixture, thus shifting the equilibrium towards the amide formation.19 Despite these early works showing great promise, direct thermal amidation has been neglected in the literature20 and is still considered limited to simple substrates.7
In the search for atom-efficient amide synthesis, we have turned to mechanochemical ball milling.21–23 Solvent-free or minimum-solvent mechanochemical reaction environment is known to have tremendous sustainability benefits24 and is known to be effective in the synthesis of amides.25–35 Here, we present a conceptually different approach that involves the thermo-mechanochemical36 treatment of carboxylic acids and amines for the quantitative conversion to amides without additives. Moreover, we observed and structurally characterised multicomponent crystalline solids as stable reaction intermediates preceding the amide bond formation. We applied our protocol for the quantitative preparation of commercial pharmaceutical amide moclobemide.
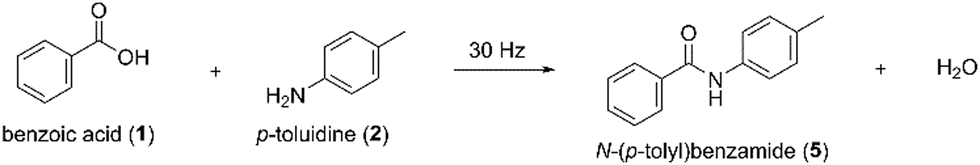
We first set out to investigate the coupling of benzoic acid (1) and p-toluidine (2). After neat grinding equimolar amounts of 1 and 2 at room temperature for 1 h (Table 1, entry 1), powder X-ray diffraction (PXRD) analysis showed the formation of a new crystalline product 3 (see ESI,† Fig. S1). However, 1H Nuclear Magnetic Resonance (1H-NMR) spectroscopy analysis showed that the amide bond formation did not occur, although there was a displacement in the chemical shift of the starting materials 1 and 2 (ESI,† Fig. S2). To better understand the origin of 3, we monitored its formation by in situ synchrotron PXRD, which showed that 3 formed within the first minute of milling and persisted until 60 min of milling (ESI,† Fig. S3). We grew single crystals of 3 and solved its crystal structure from single-crystal X-ray diffraction data (SCXRD), which revealed 3 is a cocrystal salt between ionic 1 and 2. An additional neutral molecule of 2 lies at the inversion centre, so the total 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometry in the asymmetric unit is 2:1.5 (Fig. 2a and b, Table S1 (ESI†), and ESI,† Fig. S4). The structure of 3 is characterised by a 1D hydrogen-bonded (HB) chain of R44(12) and R24(8) rings comprising the ionic 1 and 2 (Fig. 2a). The neutral molecule of 1 is attached as a pendant to this chain by a single hydrogen bond linking its OH group with a carboxylate group of anion 1 (Fig. 2a). The neutral molecule of 2 is disordered (its centroid corresponds to a crystallographic inversion centre) and does not make strong supramolecular interactions with the other crystal constituents (Fig. 2b, purple spacefill).
:2 stoichiometry in the asymmetric unit is 2:1.5 (Fig. 2a and b, Table S1 (ESI†), and ESI,† Fig. S4). The structure of 3 is characterised by a 1D hydrogen-bonded (HB) chain of R44(12) and R24(8) rings comprising the ionic 1 and 2 (Fig. 2a). The neutral molecule of 1 is attached as a pendant to this chain by a single hydrogen bond linking its OH group with a carboxylate group of anion 1 (Fig. 2a). The neutral molecule of 2 is disordered (its centroid corresponds to a crystallographic inversion centre) and does not make strong supramolecular interactions with the other crystal constituents (Fig. 2b, purple spacefill).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 (equiv.) | 2 (equiv.) | Milling temperature [°C] | Total milling time [h] | Conversion to 5a [%] |
| Reaction conditions: benzoic acid (1) (1 equiv.; 73 mg; 0.6 mmol or 2 equiv.; 147 mg; 1.2 mmol) and p-toluidine (2) (1 equiv.; 64 mg; 0.6 mmol or 2 equiv.; 129 mg; 1.2 mmol) were milled in a 12 mL stainless steel milling jar using two 1.4 g milling balls of the same material.a Determined by 1H NMR spectroscopy.b After 1 h, the milling jar was opened until cooled to room temperature then the milling was restarted for 1 h.c After 1 h, the milling jar was opened until cooled to room temperature then the milling was restarted for 2 h. | |||||
| 1 | 1 | 1 | R.t. | 1 | 0 |
| 2 | 1 | 1 | 100 | 1 | 3 |
| 3 | 1 | 1 | 160 | 1 | 25 |
| 4 | 1 | 1 | 190 | 1 | 38 |
| 5 | 1 | 1 | 190 | 3 | 58 |
| 6 | 1 | 1 | 190 | 2b | 80 |
| 7 | 1 | 1 | 190 | 3c | 89 |
| 8 | 2 | 1 | 190 | 1 | 52 |
| 9 | 1 | 2 | 190 | 1 | 11 |
| 10 | 2 | 1 | 190 | 2 | 98 |
 | ||
| Fig. 2 (a) Fragment of the main 1D HB chain in the crystal structure of 3. (b) Fragment of the crystal structure of 3 viewed down the crystallographic b axis with the disordered neutral 2 molecules depicted as purple spacefill. | ||
Milling 1 and 2 in a 2:1 stoichiometric ratio resulted in a different crystalline product 4 according to the PXRD analysis (ESI,† Fig. S1). SCXRD data (ESI,† Table S2) showed that 4 is a cocrystal salt between 1 and 2 in a 2:1 stoichiometry (the asymmetric unit contains, same as in 3, a neutral 1, and anionic 1 and cationic 2; ESI,† Fig. S5). Also, the main 1D HB chain motif of R44(12) and R24(8) rings involving ammonium cations and carboxylate anions is analogous to the chains in 3. Neutral molecules of 1 are also hydrogen-bonded as a pendant to the chains. To test other stoichiometries, we performed neat grinding of 1 and 2 in 3:1 and 1:2 ratios at room temperature for 1 h.
However, neat grinding resulted in cocrystal salts 3 or 4, respectively (ESI,† Fig. S1). To attempt the amide bond formation between 1 and 2, we employed thermo-mechanochemical reaction conditions with simultaneous mechanical and thermal activation.36,37 Recently, this strategy has been successful in obtaining amides and oligopeptides.29,38 We used a protocol in which the stainless-steel jar is equipped with a heater and controlled by a proportional-integral-derivative device for precise temperature control.29 Encouragingly, after milling equimolar amounts of 1 and 2 for 1 h at 100 °C, 3% of amide 5 was detected by 1H NMR (Table 1, entry 2). Raising the milling temperature to 160 °C resulted in 25% conversion to 5 (Table 1, entry 3), while milling at 190 °C gave 38% conversion to 5 (Table 1, entry 4).
Prolonging the milling to 3 h at 190 °C resulted in a 58% conversion to 5 (Table 1, entry 5). Being aware that removing the water from the reaction mixture enhances the amide19 and imide bond formation,39 we decided to open the hot jar after milling 1 and 2 for 1 h at 190 °C. We then waited for the milling jar to cool to room temperature and milled the same reaction mixture for an additional 1 h at 190 °C (Table 1, entry 6). This simple alteration improved the conversion to 5 to 80%. In another experiment, we lengthened the second milling cycle to 2 h, which increased the conversion to 5 up to 89% (Table 1, entry 7). We used the latter reaction conditions for scaling the reaction to a gram scale. Specifically, we milled 8.5 mmol (1.0 g) of 1 and 8.5 mmol (0.9 g) of 2 under these reaction conditions. Analysis by 1H NMR spectroscopy showed a 92% conversion to 5 (ESI,† Fig. S6). Finally, after isolation by column chromatography and purification by recrystallisation (details can be found in ESI†), we isolated 1.53 g of 5 in 85% yield.
Next, inspired by the crystal structure of cocrystal salt 4, we performed a milling experiment with 1 and 2 in a 2:1 ratio for 1 h at 190 °C (Table 1, entry 8). Surprisingly, this resulted in 52% conversion to 5, a higher conversion rate than milling equimolar amounts of 1 and 2 under the same conditions (Table 1, entry 4). On the other hand, thermo-milling 1 and 2 in a 1:2 ratio favouring an excess of 2 (Table 1, entry 9) resulted in an 11% conversion to 5. Moreover, thermo-milling 1 and 2 in a 2:1 ratio for overall 2 h (Table 1, entry 10) gave almost quantitative conversion to 5. Hence, there was a clear benefit of having an excess of 1 to achieve complete amine consumption and formation of the amide 5. To determine if cocrystal salt 4 acted as an intermediate before the amide bond formation, a milling experiment of 1 and 2 in a 2:1 ratio at 190 °C was stopped after 3 min of milling. Indeed, PXRD analysis confirmed the presence of 4 and ascertained it as a reaction intermediate. Moreover, only heating of 4 induced amide bond formation, in a higher conversion rate than heating of pristine reactants (ESI† oven heating).
Interestingly, H. Charville et al. proposed a mechanism for direct uncatalysed amidation, which involves a nucleophilic attack of an amine to one carboxylic acid molecule from a carboxylic acid hydrogen-bonded dimer.40 They proposed a concerted proton transfer mechanism in which a second carboxylic acid molecule acts as a proton acceptor from which the water molecule is eliminated. In the case of intermediates 3 and 4, the carboxylic acid units do not form hydrogen-bonded dimers but interact through single O–H⋯O hydrogen bonds (ESI,† Fig. S7). Also, the mechanism proposed by H. Charville et al. includes neutral species without an ammonium carboxylate formation. Still, the similarity to cocrystal salts 3 and 4 is in additional carboxylic acid molecule close to the reaction centre where the amide bond eventually forms.
Under high-temperature milling conditions, the reaction mixture containing 1 and 2 melts, and the amide bond formation proceeds in a molten phase. We verified this by opening the hot jar and by differential scanning calorimetry (DSC) analysis, which showed that cocrystal salts 3 and 4 melt around 53 °C and 54 °C, respectively (ESI,† Fig. S8 and S9). Additionally, we wondered if the amide-forming reaction was reversible under mechanochemical reaction conditions. We performed liquid-assisted grinding of amide 5 with H2O for 1 h, at room temperature and 120 °C. However, under these conditions, we did not observe the hydrolytic cleavage of the amide bond, and amide 5 persisted as a crystalline solid in the mixture (ESI,† Fig. S10 and S11).
We then turned to the direct mechanosynthesis of the commercially important active pharmaceutical ingredient moclobemide (9, Fig. 3a), marketed in more than 50 countries worldwide.41 Mechanosynthesis of moclobemide had previously been reported by ball milling33 and by extrusion,35 although in both cases, activated substrates (esters)33 or coupling reagents35 were needed for the amide bond formation to occur. After milling an equimolar mixture of 6 and 7 for 1 h at room temperature, we observed the formation of a new crystalline product 8, as confirmed by PXRD analysis (ESI,† Fig. S12). We solved the crystal structure of 8 from SCXRD data and revealed the formation of an ammonium carboxylate salt between 6 and 7 in 1:1 stoichiometry without neutral coformer molecules (Fig. 3b, ESI,† Table S3 and Fig. S13). Two symmetry-independent ions of 6 and 7 form two symmetry-independent 1D chains (designated as A and B) along the crystallographic b axis of the structure stabilised by strong N–H⋯O bonds (Fig. 3b and ESI,† Fig. S14). The chain A is comprised of R44(12) and R24(8) rings analogous to those in 3 and 4, while the chain B is formed by a single type of R34(10) ring. Subjecting a mixture of 6 and 7 to two thermo-milling cycles for 1 h at 190 °C resulted in a quantitative conversion to moclobemide 9 (Fig. 3a). PXRD analysis of the crude reaction mixture confirmed the formation of 9 and its crystal phase purity (ESI,† Fig. S12).
 | ||
| Fig. 3 (a) Two-step strategy for the direct thermo-mechanochemical synthesis of moclobemide 9. (b) Fragments of the supramolecular 1D HB chains A and B characteristic for the structure of the intermediate 8. The morpholine part of 7 was omitted in chain B for clarity reasons. | ||
We also milled a mixture of 6 and 7 in 2:1 stoichiometry for 1 h at room temperature, which resulted in the formation of a poorly crystalline product that was likely a mixture of phases (ESI,† Fig. S15). Nevertheless, subjecting this mixture to two thermo-milling cycles for 1 h at 190 °C also resulted in a quantitative conversion to 9. Finally, we calculated atom economy (AE) and environmental factor (E-factor) and demonstrated that thermo-mechanochemical amidation is atom-efficient (for comparison with other approaches, see ESI† green chemistry metrics). It accounts for 92.1% AE and 0.27 E-factor for 5 (ESI,† Fig. S19), and 93.7% AE and 0.07 E-factor for 9 (ESI,† Fig. S20). Importantly, model reactions between 1 and cyclohexanamine (10), and 6 and 2 led to the synthesis of amides (ESI,† synthetic procedures).
In summary, we developed a new protocol for direct amidation that involves solvent-free grinding of carboxylic acids and amines at elevated temperatures and avoids activators and additives, giving water as the only byproduct of the reaction. We observed and isolated the (cocrystal) salts between the reactants that precede the formation of amides. Although reports of non-covalent interactions preceding covalent bond formation in mechanochemistry are scarce and the phenomenon is not yet well understood,42–45 it is becoming obvious they could be involved in guiding many mechanochemical reactions. In the present work, the critical step for the high conversion to amides is milling under high temperatures and eliminating water from the reaction mixtures. This two-step thermo-mechanochemical methodology enabled the quantitative synthesis of phase pure API moclobemide 9. We will next broaden the scope of reagents to ascertain the generality of the presented methodology.
We acknowledge DESY, PETRA III (Hamburg, Germany) for providing experimental facilities and thank Dr Martin Etter for assistance in using beamline P02.1. Beamtime was allocated for proposal I-20220019 EC. K. U. acknowledges the Croatian Science Foundation IP-2020-02-4702 for financial support. We thank Dr Natalija Pantalon Juraj and Dr Aleksandar Višnjevac for help.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714 RSC.
- P. Orrechia, D. S. Petkova, R. Goetz, F. Rominger, A. S. K. Hashmi and T. Schaub, Green Chem., 2021, 23, 8169 RSC.
- K. Marchildon, Macromol. React. Eng., 2011, 5, 22 CrossRef CAS.
- A. W. Dombrowski, A. L. Aguirre, A. Shrestha, K. A. Sarris and Y. Wang, J. Org. Chem., 2022, 87, 1880 CrossRef CAS PubMed.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaksh and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140 CrossRef CAS.
- K. Ishihara, S. Ohara and H. Yamamoto, J. Org. Chem., 1996, 61, 4196 CrossRef CAS PubMed.
- H. Noda, M. Furutachi, Y. Asada, M. Shibasaki and N. Kumagai, Nat. Chem., 2017, 9, 571 CrossRef CAS PubMed.
- M. T. Sabatini, L. T. Boulton and T. D. Sheppard, Sci. Adv., 2017, 3, e1701028 CrossRef PubMed.
- C. L. Allen, A. R. Chhatwala and J. M. J. Williams, Chem. Commun., 2012, 48, 666 RSC.
- H. Lundberg, F. Tinnis and H. Adolfsson, Chem. – Eur. J., 2012, 18, 3822 CrossRef CAS PubMed.
- H. Lundberg, F. Tinnis and H. Adolfsson, Synlett, 2012, 2201 CAS.
- H. Lundberg and H. Adolfsson, ACS Catal., 2015, 5, 3271 CrossRef CAS.
- M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, Nat. Catal., 2019, 2, 10 CrossRef CAS.
- A. G. von Hofmann, Ber. Dtsch. Chem. Ges., 1882, 15, 977 CrossRef.
- J. A. Mitchell and E. E. Reid, J. Am. Chem. Soc., 1931, 53, 1879 CrossRef CAS.
- J. Cossy and C. Pale-Grosdemange, Tetrahedron Lett., 1989, 30, 2771 CrossRef CAS.
- B. S. Jursic and Z. Zdravkovski, Synth. Commun., 1993, 23, 2761 CrossRef CAS.
- L. J. Gooßen, D. M. Ohlmann and P. P. Lange, Synthesis, 2009, 160 CrossRef.
- S. L. James, et al. , Chem. Soc. Rev., 2012, 41, 413 RSC.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018 CrossRef PubMed.
- A. A. L. Michalchuk, E. V. Boldyreva, A. M. Belenguer, F. Emmerling and V. V. Boldyrev, Front. Chem., 2021, 9, 685789 CrossRef CAS PubMed.
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145 CrossRef CAS PubMed.
- T.-X. Metro, J. Bonnamour, T. Reidon, J. Sarpoulet, J. Martinez and F. Lamaty, Chem. Commun., 2012, 48, 11781 RSC.
- V. Štrukil, B. Bartolec, T. Portada, I. Đilović, I. Halasz and D. Margetić, Chem. Commun., 2012, 48, 12100 RSC.
- A. Wróblewska, P. Paluch, E. Wielgus, G. Bujacz, M. K. Dudek and M. J. Potrzebowski, Org. Lett., 2017, 19, 5360 CrossRef PubMed.
- C. Duangkamol, S. Jaita, S. Wangngae, W. Phakhodee and M. Pattarawarapan, RSC Adv., 2015, 5, 52624 RSC.
- N. Cindro, M. Tireli, B. Karadeniz, T. Mrla and K. Užarević, ACS Sustainable Chem. Eng., 2019, 7, 16301 CrossRef CAS.
- T. Dalidovich, K. A. Mishra, T. Shalima, M. Kudrjašova, D. G. Kananovich and R. Aav, ACS Sustainable Chem. Eng., 2020, 8, 15703 CrossRef CAS.
- E. Broumidis, M. C. Jones, F. Vilela and G. O. Lloyd, ChemPlusChem, 2020, 85, 1754 CrossRef CAS PubMed.
- J. Gómez-Carpintero, J. D. Sánchez, J. F. González and J. C. Menéndez, J. Org. Chem., 2021, 86, 14232 CrossRef PubMed.
- W. Nicholson, F. Barreteau, J. A. Leitch, R. Payne, I. Priestley, E. Godineau, C. Battilocchio and D. L. Browne, Angew. Chem., Int. Ed., 2021, 60, 21868 CrossRef CAS PubMed.
- R. Mocci, E. Colacino, L. De Luca, C. Fattuoni, A. Porcheddu and F. Delogu, ACS Sustainable Chem. Eng., 2021, 9, 2100 CrossRef CAS.
- M. Lavayssiere and F. Lamaty, Chem. Commun., 2023, 59, 3439 RSC.
- V. Martinez, T. Stolar, B. Karadeniz, I. Brekalo and K. Užarević, Nat. Rev. Chem., 2023, 7, 51 CrossRef CAS PubMed.
- G. Felix, N. Fabregue, C. Leroy, T.-X. Metro, C.-H. Chen and D. Laurencin, Phys. Chem. Chem. Phys., 2023, 25, 23435 RSC.
- T. Stolar, S. Grubešić, N. Cindro, E. Meštrović, K. Užarević and J. G. Hernández, Angew. Chem., Int. Ed., 2021, 60, 12727 CrossRef CAS PubMed.
- T. Rensch, S. Fabig, S. Grätz and L. Borchardt, ChemSusChem, 2022, 15, e202101975 CrossRef CAS PubMed.
- H. Charville, D. A. Jackson, G. Hodges, A. Whiting and M. R. Wilson, Eur. J. Org. Chem., 2011, 5981 CrossRef CAS.
- D. T. Chen and R. Ruch, Clin. Neuropharmacol., 1993, 16, S63 Search PubMed.
- S. Lukin, M. Tireli, I. Lončarić, D. Barišić, P. Šket, D. Vrsaljko, M. di Michiel, J. Plavec, K. Užarević and I. Halasz, Chem. Commun., 2018, 54, 13216 RSC.
- M. Kralj, S. Lukin, G. Miletić and I. Halasz, J. Org. Chem., 2021, 86, 14160 CrossRef CAS PubMed.
- K. J. Ardila-Fierro, M. Rubčić and J. G. Hernández, Chem. – Eur. J., 2022, 28, e202200737 CrossRef CAS PubMed.
- J. G. Hernández, K. J. Ardila-Fierro, S. Gómez, T. Stolar, M. Rubčić, E. Topić, C. Z. Hadad and A. Restrepo, Chem. – Eur. J., 2023, e202301290 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2287225, 2287226 and 2285230. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc04448c |
| ‡ Current address: Federal Institute for Materials Research and Testing (BAM), 12489 Berlin, Germany. |
| This journal is © The Royal Society of Chemistry 2023 |