
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isolation of the elusive [Ru(bipy)3]+: a key intermediate in photoredox catalysis†
Samuel J.
Horsewill
 a,
Chengyang
Cao
b,
Noah
Dabney
c,
Eric S.
Yang
b,
Stephen
Faulkner
b and
Daniel J.
Scott
*a
a,
Chengyang
Cao
b,
Noah
Dabney
c,
Eric S.
Yang
b,
Stephen
Faulkner
b and
Daniel J.
Scott
*a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: ds2630@bath.ac.uk
bDepartment of Chemistry, Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK
cDepartment of Chemistry, Guilford College, 5800 W Friendly Ave., Greensboro, NC 27410, USA
First published on 15th November 2023
Abstract
Photoredox catalysis has flourished in recent years, but due to its widespread utility applications have grown faster than mechanistic understanding. In this report we help to address this deficit by isolating and characterising one of the intermediates of the iconic photocatalyst [Ru(bipy)3]2+, and testing its initial photoreactivity towards common substrates.
Ruthenium bipyridyl complexes have played a key role in the development of the field of photoredox catalysis (PRC) since its inception 40 years ago.1 Notably, in 2008 the groups of MacMillan and Yoon pioneered the application of visible light PRC to organic transformations using [Ru(bipy)3]Cl2 ([Ru]Cl2) as a photocatalyst, helping to spark the recent surge of interest in synthetic PRC.2 Since then the field has expanded rapidly to the point that PRC has become a key tool in the arsenal of synthetic organic chemists.3 Due to its established vital role, [Ru]Cl2 continues to be widely employed in PRC and is often used as a “model” system.4 The concept behind PRC is simple: a photocatalyst PC is excited by a photon, and in the resulting excited state has a higher reduction and/or oxidation potential (Fig. 1a, pathway I). It is then able to donate or accept an electron to or from a substrate (oxidative or reductive quenching, respectively), in turn leading to downstream product formation. Using PRC, even challenging substrates and reactions which were previously performed using forcing conditions can now be performed in mild, safe procedures.5 However, in reality mechanisms are rarely as simple as this basic picture suggests, frequently featuring several interacting cycles, multiple radical intermediates, and transient reactive species.6
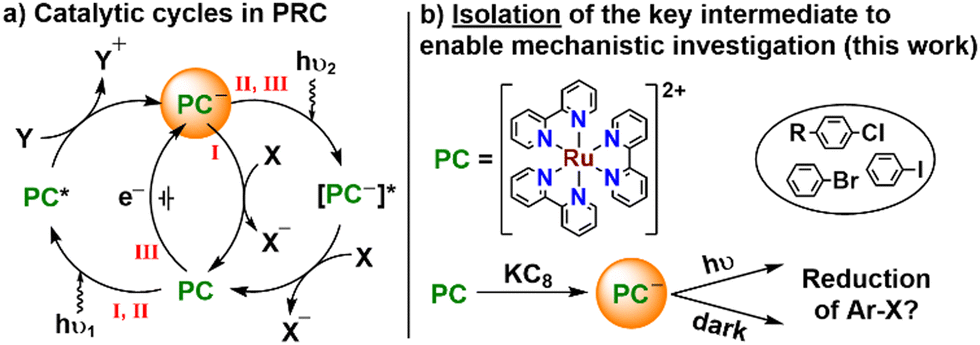 | ||
| Fig. 1 (a) General scheme for reductive quenching in photoredox catalysis (PRC), showing pathways for I “traditional” PRC, II ConPET and III e-PRC; (b) Our strategy of isolation of reactive intermediates to enable mechanistic understanding. PC = photocatalyst; X = oxidative quencher; Y = reductive quencher. | ||
In particular, more complex mechanisms involving the reactivity of radical excited states in reductive consecutive photoelectron transfer (ConPET) and electron-primed PRC (e-PRC) have recently been the subject of significant controversy (Fig. 1a, pathways II and III).7 Both of these mechanisms involve electron transfer from radical excited states of a photocatalyst, *PC˙−, that are known to be very short-lived, leading some to question their viability.7g However, these excited states have the exceptional redox strength required to reduce very challenging substrates (as low as ca. −3 V vs. SCE), which are shown to be activated under these conditions.8 While they have not yet been reported to react by exactly the same ConPET/e-PRC mechanisms, similar reactivity has also been described for ruthenium polypyridyl complexes. Specifically, for some complexes including [Ru]2+, after accepting an electron from a donor the one-electron reduced complex [Ru]+ is reported to be able to undergo a second photoexcitation to form a highly reducing excited state, *[Ru]+.9 This excited state is then suggested to release a free, solvated electron as a potent and relatively long-lived reductant which can go on to reduce challenging substrates such as aryl chlorides. A similar mechanism has also been proposed in other PRC systems as an alternative to radical excited state mechanisms.10 However, the feasibility of many of these processes remains controversial, despite recent attempts at clarification.11–13
Given the intense research interest in and open questions around PRC, it is surprising that until recently, detailed investigation of its underlying mechanisms has not commonly been undertaken.14 In only a few cases has isolation of the proposed reactive intermediates in oxidative11 and reductive12,13 quenching cycles been reported. And it is only this year that we and the group of Knowles have isolated the first examples of one-electron reduced intermediates of organic and iridium-based photocatalysts, respectively, which provided direct insight into controversial ConPET and e-PRC mechanisms. Remarkably, to our knowledge no such effort has yet been reported for ruthenium photocatalysts, despite their prominence as the “model” PRC system. This is particularly surprising given the ubiquitous applications of [Ru]2+ and related ruthenium polypyridyl complexes in solar cells, LEDs, and many other areas of photochemistry, as well as PRC.15 We were therefore motivated to extend our previous work by targeting 1e− reduction of this iconic PC to obtain the key reactive intermediate in the PRC reductive quenching cycle of [Ru]2+, i.e. [Ru]+, which we describe herein. There have been previous reports of the electrochemical isolation of [Ru]n[C60]m (n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :m = 1:2, 1:1, 2:1), where [Ru]2[C60] is proposed to contain the [Ru]+ cation.16 However, in that case it is difficult to decouple the contributions of [Ru]+ from the electronically complex and redox non-innocent fullerene. By chemical reduction of the iconic ruthenium photocatalyst [Ru]2+ we have instead isolated the widely proposed intermediate [Ru]+ as a “bottleable” material with an inert counter-anion, [Ru][BPh4]. We have characterised [Ru][BPh4] spectroscopically and structurally, and performed some preliminary studies of its reactivity with aryl halides to shed initial light on its photoredox reactivity.
:m = 1:2, 1:1, 2:1), where [Ru]2[C60] is proposed to contain the [Ru]+ cation.16 However, in that case it is difficult to decouple the contributions of [Ru]+ from the electronically complex and redox non-innocent fullerene. By chemical reduction of the iconic ruthenium photocatalyst [Ru]2+ we have instead isolated the widely proposed intermediate [Ru]+ as a “bottleable” material with an inert counter-anion, [Ru][BPh4]. We have characterised [Ru][BPh4] spectroscopically and structurally, and performed some preliminary studies of its reactivity with aryl halides to shed initial light on its photoredox reactivity.
First, we attempted the reduction of commercially available [Ru]Cl2. Reaction with KC8 resulted in an immediate colour change to deep red/pink, and isolation of the products via Soxhlet extraction into THF provided the target [Ru]Cl in 25% yield. The low yield is attributed to very low solubility in even THF. Following recent reports suggesting that [Ru]+ can be an active intermediate in water,9b we considered that this could be an appropriate alternative solvent for the extraction. However, decomposition back to [Ru]2+ was observed when a small-scale reaction was extracted into water, indicating that [Ru]+ is only transiently stable under aqueous conditions. Thus, a species was required that would instead be more soluble in non-aqueous solution. The tetraphenyl borate anion [BPh4]− was identified as an appropriate outer sphere counter ion to impart this solubility. Following reduction of [Ru]Cl2 with KC8, 1 eq. Na(BPh4) was added prior to Soxhlet extraction of the product, providing [Ru][BPh4] in a significantly improved 54% yield as shown in Fig. 2a. As isolated in this manner, small quantities of silicone grease remained in the product, identified by 1H NMR spectroscopy. However, recrystallisation from saturated MeCN solution at −35 °C provided analytically pure [Ru][BPh4]·MeCN. Apart from the [BPh4]− resonances, a single broad, attenuated resonance was detected in the 1H NMR spectrum of [Ru][BPh4] (δ = 23.41 ppm), assigned to the bipy ligands. This is consistent with the expected open–shell character. Elemental analysis was consistent with the expected composition (see ESI,† Section 2.3).
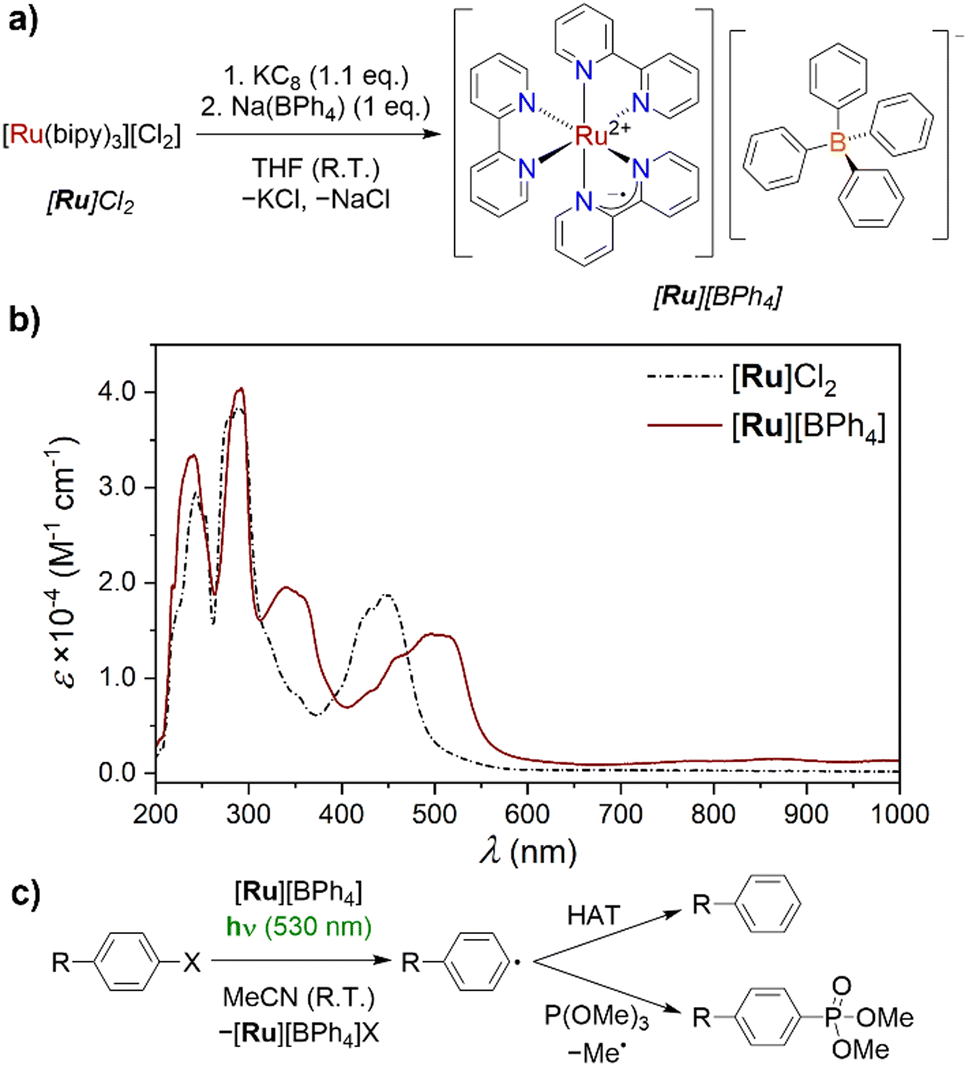 | ||
| Fig. 2 (a) Scheme illustrating the reduction of [Ru]Cl2 and synthesis of [Ru][BPh4]. (b) UV-vis absorption spectrum of [Ru]Cl2, recorded on a 520 μM MeCN solution, and [Ru][BPh4], recorded on a 500 μM MeCN solution. Both spectra were recorded at room temperature in a 1 mm cuvette. (c) The model reaction used to investigate photoreactivity of [Ru][BPh4], showing the primary products. R = H, OMe, CO2Me; X = Cl, Br, I. | ||
The X-band EPR spectrum of [Ru][BPh4] (see ESI,† Fig. S10) was recorded at room temperature. The spectrum obtained exhibited a single, broad resonance at g = 2.002, consistent with an organic-centred radical, and hence reduction being localised on the bipy ligands (vide infra). While not a perfect comparison due to temperature differences and the absence of electrolyte, the broad linewidth is consistent with previously conducted EPR studies on electrochemically generated [Ru]+, and has been attributed to pseudo-spin-rotational coupling.17
The UV-vis absorption spectrum of [Ru][BPh4] (Fig. 2b) exhibits a series of intense transitions in both the visible and UV regions of the spectrum, as expected for a species containing an organic radical, and is consistent with spectra recorded in situ on electrochemically generated [Ru]+.18 The dominant feature in the visible region is a broad envelope between 450 and 550 nm, at lower energy compared to that of [Ru]Cl2, which exhibits its greatest maximum at 445 nm. A transition with a maximum at 340 nm, as well as some low-intensity absorptions extending into the NIR region also grow in following reduction. These have been attributed to π–π* transitions of the bipy ligands.18b (The expected inter-valence charge transfer band was previously identified outside our spectral window, at 2200 nm).18c [Ru]Cl exhibits similar absorptions to [Ru][BPh4] (see ESI,† Fig. S5).
Diffusion of hexanes into a saturated THF solution of [Ru][BPh4] produced X-ray diffraction (XRD) quality single crystals, providing the solid-state molecular structure (SSMS) of [Ru][BPh4]·THF shown in Fig. 3. In this structure, [Ru]+ and the [BPh4]− counter-anion form a separated ion pair, co-crystallising with one molecule of THF solvent. Previously, well-defined complexes with discrete, unambiguously defined counter ions containing a [Ru]n+ unit have been structurally characterised for n = 3, 2 and 0 but not for n = 1. [Ru][BPh4]·THF was therefore compared to selected examples ([Ru][PF6]3,19 [Ru][PF6]2,20 and [Ru]0;21 full bond information in the ESI,† Table S1 and S2).
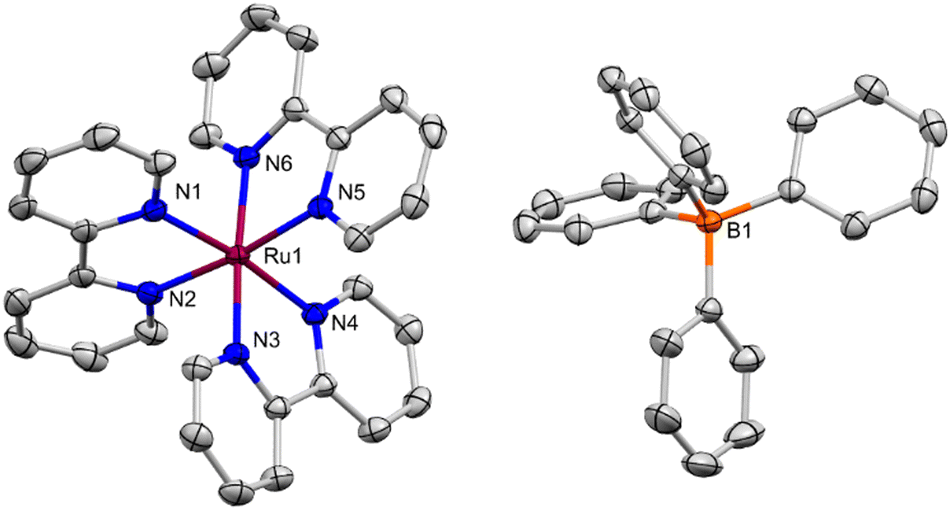 | ||
| Fig. 3 Solid-state molecular structure of [Ru][BPh4]·THF, crystallised by diffusion of hexanes into a saturated THF solution. Thermal ellipsoids are drawn at 50% probability. H atoms and a THF molecule are omitted for clarity. Atom colours: Ru, burgundy; N, blue; B, orange; C, grey. | ||
The C2–C2′ bond linking the two pyridyl rings of the bipy ligand (Fig. S50, ESI†) is particularly diagnostic of its oxidation state. Notably, in [Ru][BPh4]·THF, the bipy ligands are non-equivalent. Two of the ligands feature C2–C2′ bonds (1.464(3)–1.473(2) Å) indistinguishable from those observed in [Ru][PF6]2 (1.470(6)–1.478(5) Å), consistent with a neutral bipy formulation.20 However, the other bipy ligand exhibits a much shorter bond (1.418(3) Å), consistent with addition of an electron to a π* orbital, and hence reduction to bipy˙− (see ESI,† Fig. S50). This bond distance is more similar to the bonds in [Ru]0 (1.431(5)–1.448(4) Å).21 No close, non-packing interactions of the bipy ligands with the [BPh4]− or THF molecule were identified. It seems therefore that in the SSMS of [Ru][BPh4]·THF, the unpaired electron is localised onto a single bipy ligand, with overall formulation as [Ru2+(bipy)2(bipy˙−)]. The related [Cr(tbipy)2(tbipy˙−)]2+ (tbipy = 4,4′-Di-tbutyl-2,2′-bipyridine) has a similar localised structure.22 In surprising contrast, single-crystal XRD quality crystals grown from a saturated MeCN solution at −35 °C, which gave the similar [Ru][BPh4]·MeCN, did not exhibit analogous differences in bond distances, with the bipy ligands instead being statistically equivalent (see ESI,† Section 4.3; this is similar to [Ru]0/3+).
It is worth noting that the degree of delocalisation of the unpaired electron across the bipy ligands in [Ru]+ has been the topic of considerable discussion. Most recently, spectroscopic studies of the excited states of [Ru(bipy)n(L)3−n]2+ and the series [Ru(bipy)3]n (n = +2 to −1), alongside DFT calculations, have supported either full delocalisation, or a formally localised description where the unpaired electron hops rapidly between all three bipy sites.17,18,23 In either case the electron would be expected to be evenly distributed across the three bipy ligands over the timescale of an XRD experiment. That this is seemingly not the case for crystalline [Ru][BPh4]·THF suggests that fully localised electronic structures must be very close in energy, with crystal packing effects able to influence their population.
With [Ru][BPh4] in hand, we set about also performing a preliminary investigation into its reactivity to provide an initial demonstration of its utility as a tool for mechanistic study. To determine the solution stability of [Ru][BPh4] under experimental conditions, 2.5 mM solutions in MeCN were irradiated with LED light of three different wavelengths, λmax = 365, 455 and 530 nm, chosen to match the absorbance spectrum of [Ru][BPh4]. When irradiated for 16 hours [Ru][BPh4] exhibited gradual decomposition, as 1H NMR resonances consistent with the bipy ligands of [Ru]2+ appeared over time. Decomposition was significant when irradiated with light of either 455 or 365 nm, but with 530 nm light only trace [Ru]2+ (4%) was observed. After only 1 hour at 530 nm, no decomposition was detected, and 530 nm LEDs were therefore used for subsequent reactions.
The model reaction chosen for this initial study was the reduction of aryl halide substrates PhX (X = Cl, Br, I; Fig. 2c). Of these, PhCl is the most challenging to reduce, followed by PhBr and PhI. The reduction of such species is frequently reported in photoredox reactions,9b,24 and is used here as a model to probe the reactivity of [Ru][BPh4]. Following reduction of an aryl halide, the halide is rapidly ejected to form an aryl radical, which can go on to further react in bond-forming reactions. All reactions were performed using a 2.5 mM solution of [Ru][BPh4], with 20 eq. of aryl halide substrate and radical trap where relevant (50 mM concentration), to mimic catalytic conditions.13 Note that this concentration of [Ru][BPh4] is near the upper limit of its solubility in MeCN.
In the absence of light, [Ru][BPh4] reacted only with PhI, resulting in oxidation of the former, as evidenced by 1H NMR observation of diamagnetic [Ru]2+. However, upon irradiation at 530 nm for 1 h similar reactivity was also observed using PhBr, with only PhCl not exhibiting obvious production of [Ru]2+. The other predicted products of these reactions, benzene and biphenyl (derived from Ph˙ by H atom abstraction and homocoupling respectively), were not measured directly as their 1H NMR resonances overlap with those of PhX so could not be detected in this manner. Instead, generation of phenyl radicals was confirmed using the radical trap P(OMe)3, which reacts with phenyl radicals to produce the phosphonate PhPO(OMe)2. It was first confirmed that [Ru][BPh4] does not react with P(OMe)3 even under irradiation, then mixtures of [Ru][BPh4], PhX and P(OMe)3 were irradiated at 530 nm for 1 h. Gratifyingly, in all three reactions the characteristic 31P NMR resonance of PhPO(OMe)2 was detected, and quantified relative to a subsequently-added internal standard (Ph3PO). Curiously, the highest yield was obtained for the reaction with PhBr (60% with respect to [Ru]+), followed by PhI (26%) and PhCl (13%). We also studied alternative aryl chloride substrates functionalised with either an electron withdrawing group (MeO2C(C6H4)Cl) or an electron donating group (MeO(C6H4)Cl). The yield of phosphonate product with the EWG was higher (17%) than that for the parent PhCl, while that with the EDG was lower (4%), consistent with the different potentials required to reduce these substrates. Moreover, when the reduction of PhBr in the presence of P(OMe)3 was performed over an extended period of 16 h, a far higher, greater than stoichiometric yield of 205% was recorded. We attribute this formal turnover to reduction of [Ru]2+ back to [Ru]+ by a phosphoranyl intermediate, as observed in our recent study of organic PC˙−, suggesting that this is a fairly general process.13 Control reactions were performed using [Ru]Cl2 in place of [Ru][BPh4], and did not show the same reactivity (see ESI,† Section 3.3).
In summary, we have isolated [Ru][BPh4] as a bottleable source of [Ru]+, a proposed key intermediate in many different reactions of the ubiquitous photocatalyst [Ru]2+ and previously a conspicuous missing link in the series of isolated compounds [Ru]n+. Our investigations suggest that the unpaired electron can be surprisingly localised on a single bipy ligand, at least in the solid state. Initial reactivity studies show that [Ru]+ is itself photochemically active, which must be factored into any future analysis of [Ru]n+ photoreactivity.
The authors would like to thank the EPSRC for funding (S. J. H., D. J. S.: EP/V056069/1; E. S. Y.: EP/S023828/1), the University of Oxford for access for Chemical Crystallography facilities, and the University of Bath for technical support and assistance.25
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) D. M. Hedstrand, W. M. Kruizinga and R. M. Kellogg, Tetrahedron Lett., 1978, 19, 1255–1258 CrossRef; (b) T. J. van Bergen, D. M. Hedstrand, W. H. Kruizinga and R. M. Kellogg, J. Org. Chem., 1979, 44, 4953–4962 CrossRef CAS; (c) K. Hironaka, S. Fukuzumi and T. Tanaka, J. Chem. Soc., Perkin Trans. 2, 1984, 1705–1709 RSC; (d) C. Pac, M. Ihama, M. Yasuda and H. Sakurai, J. Org. Chem., 1984, 49, 26–34 CrossRef CAS.
- (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322(5898), 77–80 CrossRef CAS PubMed; (b) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed.
- (a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (b) M. Cybularczyk-Cecotka, J. Szczepanik and M. Giedyk, Nat. Catal., 2020, 3, 872–886 CrossRef CAS; (c) A. R. Allen, E. A. Noten and C. R. J. Stephenson, Chem. Rev., 2022, 122, 2695–2751 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- (a) J. Castellanos-Soriano, J. C. Herrera-Luna, D. D. Díaz, M. C. Jiménez and R. Perez-Ruiz, Org. Chem. Front., 2020, 7, 1709 RSC; (b) D. De Vos, K. Gadde and B. U. W. Maes, Synthesis, 2023, 193–231 CrossRef CAS; (c) S. Wu, J. Kaur, T. A. Karl, X. Tian and J. P. Barham, Angew. Chem., Int. Ed., 2022, 61, e202107811 CrossRef CAS PubMed; (d) M. Lepori, S. Schmid and J. P. Barham, Beilstein J. Org. Chem., 2023, 19, 1055–1145 CrossRef CAS PubMed.
- (a) K. Goliszewska, K. Rybicka-Jasińska, J. A. Clark, V. I. Vullev and D. Gryko, ACS Catal., 2020, 10, 5920–5927 CrossRef CAS; (b) M. S. Coles, G. Quach, J. E. Beves and E. G. Moore, Angew. Chem., Int. Ed., 2020, 59, 9522–9526 CrossRef CAS PubMed; (c) B. G. Stephenson, E. H. Spielvogel, E. A. Loiaconi, V. Mulwa Wambua, R. V. Nakhamiyayev and J. R. Swierk, J. Am. Chem. Soc., 2021, 143, 8878–8885 CrossRef PubMed.
- (a) N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2020, 142, 2093–2099 CrossRef CAS PubMed; (b) H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS PubMed; (c) J. S. Beckwith, A. Aster and E. Vauthey, Phys. Chem. Chem. Phys., 2022, 24, 568–577 RSC; (d) I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed; (e) M. Marchini, A. Gualandi, L. Mengozzi, P. Franchi, M. Lucarini, P. G. Cozzi, V. Balzani and P. Ceroni, Phys. Chem. Chem. Phys., 2018, 20, 8071–8076 RSC; (f) C. J. Zeman IV, S. Kim, F. Zhang and K. S. Schanze, J. Am. Chem. Soc., 2020, 142, 2204–2207 CrossRef PubMed; (g) A. J. Reith, M. I. Gonzalez, B. Kudisch, M. Nava and D. G. Nocera, J. Am. Chem. Soc., 2021, 143, 14352–14359 CrossRef PubMed.
- (a) F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chem., Int. Ed., 2020, 59, 10266–10284 CrossRef CAS PubMed; (b) H. Huang, K. A. Steiniger and T. H. Lambert, J. Am. Chem. Soc., 2022, 144, 12567–12583 CrossRef CAS PubMed.
- (a) R. Naumann, F. Lehmann and M. Goez, Angew. Chem., Int. Ed., 2017, 57, 1078–1081 CrossRef PubMed; (b) R. Naumann and M. Goez, Chem. – Eur. J., 2018, 24, 17557–17567 CrossRef CAS PubMed.
- (a) C. Kerzig and O. S. Wenger, Chem. Sci., 2019, 10, 11023 RSC; (b) J. P. Cole, D.-F. Chen, M. Kudisch, R. M. Pearson, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2020, 142, 13573–13581 CrossRef CAS PubMed.
- (a) S. Wu, J. Žurauskas, M. Domański, P. S. Hitzfeld, V. Butera, D. J. Scott, J. Rehbein, A. Kumar, E. Thyrhaug, J. Hauer and J. P. Barham, Org. Chem. Front., 2021, 8, 1132 RSC; (b) A. Kumar, P. Malevich, L. Mewes, S. Wu, J. P. Barham and J. Hauer, J. Chem. Phys., 2023, 158, 144201 CrossRef CAS PubMed.
- Y. Baek, A. Reinhold, L. Tian, P. D. Jeffrey, G. D. Scholes and R. R. Knowles, J. Am. Chem. Soc., 2023, 145, 12499–12508 CrossRef CAS PubMed.
- S. J. Horsewill, G. Hierlmeier, Z. Farasat, J. P. Barham and D. J. Scott, ACS Catal., 2023, 13, 9392–9403 CrossRef CAS PubMed.
- (a) A. Seegerer, P. Nitschke and R. M. Gscwind, Angew. Chem., Int. Ed., 2018, 57, 7493–7497 CrossRef CAS PubMed; (b) M. S. Coles, G. Quach, J. E. Beves and E. G. Moore, Angew. Chem., Int. Ed., 2020, 59, 9522–9526 CrossRef CAS PubMed; (c) N. A. Till, L. Tian, Z. Dong, G. D. Scholes and D. W. C. MacMillan, J. Am. Chem. Soc., 2020, 142, 15830–15841 CrossRef CAS PubMed.
- (a) D. W. Thompson, A. Ito and T. J. Meyer, Pure Appl. Chem., 2013, 85, 1257–1305 CrossRef CAS; (b) C. Förster and K. Heinze, Chem. Soc. Rev., 2020, 49, 1057 RSC.
- J. Hong, M. P. Shores and C. M. Elliott, Inorg. Chem., 2010, 49, 11378–11385 CrossRef CAS PubMed.
- (a) A. G. Motten, K. Hanck and M. K. DeArmond, Chem. Phys. Lett., 1981, 79, 541–546 CrossRef CAS; (b) D. E. Morris, K. W. Hanck and M. K. DeArmond, J. Am. Chem. Soc., 1983, 105, 3032–3038 CrossRef CAS.
- (a) S. Hohloch, D. Schweinfurth, M. G. Sommer, F. Weisser, N. Deibel, F. Ehret and B. Sarkar, Dalton Trans., 2014, 43, 4437–4450 RSC; (b) G. A. Heath, L. J. Yellowlees and P. S. Braterman, J. Chem. Soc., Chem. Commun., 1981, 287–289 RSC; (c) G. A. Heath, L. J. Yellowlees and P. S. Braterman, Chem. Phys. Lett., 1982, 92, 646–648 CrossRef CAS.
- M. Biner, H.-B. Bürgi, A. Ludi and C. Röhr, J. Am. Chem. Soc., 1992, 114, 5197–5203 CrossRef CAS.
- J. Breu, H. Domel and A. Stoll, Eur. J. Inorg. Chem., 2000, 2401–2408 CrossRef CAS.
- E. E. Pérez-Cordero, C. Campana and L. Echegoyen, Angew. Chem., Int. Ed. Engl., 1997, 36, 137–140 CrossRef.
- C. C. Scarborough, S. Sproules, T. Weyhermüller, S. DeBeer and K. Wieghardt, Inorg. Chem., 2011, 50, 12446–12462 CrossRef CAS PubMed.
- (a) J. England, C. C. Scarborough, T. Weyhermüller, S. Sproules and K. Wieghardt, Eur. J. Inorg. Chem., 2012, 4605–4621 CrossRef CAS; (b) A. Suris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef.
- (a) P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084 CrossRef CAS PubMed; (b) K. R. Howes, A. Bakac and J. H. Espenson, Inorg. Chem., 1988, 27, 3147 CrossRef CAS; (c) J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- Material and Chemical Characterisation Facility (MC2), University of Bath DOI:10.15125/mx6j-3r54.
Footnote |
| † Electronic supplementary information (ESI) available: Further experimental and synthetic details, NMR spectra and structure data. CCDC 2290574 and 2290732. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc04375d |
| This journal is © The Royal Society of Chemistry 2023 |