
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of Li1.20Mn0.432+Nb0.39O2 disordered rock-salt under reducing conditions for Li-ion batteries†
Wilgner Lima
da Silva
 ab,
Ashok S.
Menon
b,
Martin R.
Lees
c,
Reza J.
Kashtiban
c,
Marc
Walker
c,
Louis F. J.
Piper
b,
Emma
Kendrick
*d and
Richard I.
Walton
*a
ab,
Ashok S.
Menon
b,
Martin R.
Lees
c,
Reza J.
Kashtiban
c,
Marc
Walker
c,
Louis F. J.
Piper
b,
Emma
Kendrick
*d and
Richard I.
Walton
*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: r.i.walton@warwick.ac.uk
bWMG, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK
cDepartment of Physics, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK
dSchool of Metallurgy and Materials, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: e.kendrick@bham.ac.uk
First published on 19th October 2023
Abstract
A Mn2+-Li-Nb disordered rock-salt oxide cathode is prepared by a solid-state reaction under 5% H2/N2, and its electrochemical property shows a high voltage plateau at 4.8 V, with irreversible structural changes in the 1st cycle due to O redox processes; this is supported by powder X-ray diffraction and ex situ laboratory Mn K-edge XANES data.
Searching for new cathode materials is a crucial step to reach high energy density positive electrodes for the next generation of Li-ion batteries. Disordered rock-salt (DRS) cathodes are a promising class of positive electrode materials due to their high specific capacity (≥300 mA h g−1) and energy density1–3 compared to conventional layered rock-salt derived layered nickel-manganese-cobalt (NMC) oxides. However, DRS positive electrodes suffer from poor capacity retention, large voltage hysteresis and oxygen redox activity at high voltages that are detrimental to their long term behaviour.4–6
The common synthesis approaches to DRS oxides are solid-state reactions under Ar, N2 or air, and mechanochemical synthesis routes. These allow access to transition-metal niobates (Li,Nb,M)O with M = Mn3+, Fe3+, Co3+, V3+etc.5–7 To modify the transition-metal oxidation state and to moderate oxygen redox, partial replacement of oxide by fluoride in this material is an important strategy.1,8,9 This allows access to materials with a lower transition-metal oxidation state, such as Mn2+, to adjust electrochemical performance. Solid-state synthesis forms the most thermodynamically stable phases, whilst mechanochemical synthesis generates defects and amorphous species without heat treatment,5,8 but its application is hindered in large-scale manufacturing purposes because of irreproducibility and impurities from the grinding media.10 In this paper, we report a different approach to access low oxidation states of transition-metal cations in lithium niobate DRS materials, with the use of a reductive gas atmosphere during synthesis.
Li1.20Mn0.43Nb0.39O2 (LMNbO) was prepared by a solid-state synthesis at 900 °C for 6 hours under flowing 5% H2/N2 following roll-milling and pelletisation of oxide precursors (see ESI†). Only this composition was found to be accessible under the conditions used, Fig. S2 (ESI†), and even under a N2 atmosphere the product contains some Mn3+ (Fig. S6, ESI†)
Mn K-edge X-ray absorption near edge structure (XANES) spectra (B18, Diamond Light Source) revealed that the as-made sample contains Mn with an oxidation state of 2+, Fig. 1a, by comparison to reference Mn-containing materials, Fig. 1b. Fig. 1c shows the Pawley fit of high resolution powder X-ray diffraction data (I11, Diamond Light Source) of LMNbO using the ternary rock-salt Li0.578Nb0.294O as a starting model,11 and Table 1 gives the cubic unit cell parameter compared to other relevant materials. Another plot of the fitted powder X-ray diffraction (PXRD) data is shown in Fig. S3 (ESI†). A model with more than one cubic unit cell was necessary to give a satisfactory fit, which is evident in the PXRD peak shape anisotropy. The Pawley refinement confirmed that LMNbO sample has a larger unit cell compared to Li1.25Mn0.253+Nb0.50O2, and this is expected due to the larger ionic radius of Mn2+ (0.83 Å)12 compared to Mn3+ (0.645 Å).12 It should be noted that Li1.25Mn3+0.25Nb0.50O2 has a different space group as a consequence of the presence of domains of order, which were detected using total scattering analysis.13 Domains of order appear as a broad peak at low 2θ values, present in the as-made LMNbO, Fig. S5 (ESI†).
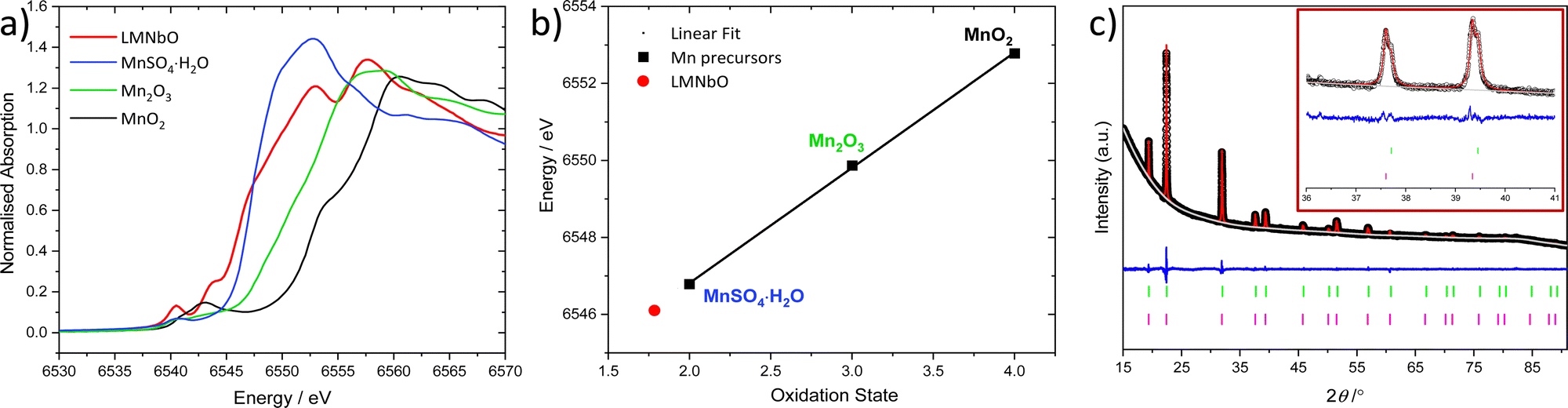 | ||
| Fig. 1 (a) Mn K-edge XANES spectra of as-made LMNbO and Mn standards (MnSO4·H2O (blue line), Mn2O3 (green line) and MnO2 (black line)), and (b) linear fit of edge energy (0.5 normalised absorption) to define the oxidation state of Mn in LMNbO, (c) Pawley refinement fit of PXRD data [λ = (0.826858 ± 0.00206(6)) Å] of as-made LMNbO (Rwp = 1.43%, GOF = 2.15) and insert depicts high 2θ values (experimental data (black), fitted profile (red), background (grey), difference (blue), allowed Bragg peaks (green and pink)). | ||
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, and Li1.25Mn0.253+Nb0.50O2 space group: I41/amd with the cubic equivalent lattice parameter given)
m, and Li1.25Mn0.253+Nb0.50O2 space group: I41/amd with the cubic equivalent lattice parameter given)
Energy-dispersive X-ray spectroscopy (EDS) maps measured using transmission electron microscopy (TEM) (Fig. 2) showed homogeneous elemental distribution of Nb and Mn within particles over the range of tens of nanometres, and the elemental composition from scanning electron microscopy (SEM) coupled with EDS maps (Fig. S7, ESI†) and X-ray fluorescence (XRF) (Table S3, ESI†) measurements are similar and near the expected values for both transition metals in LMNbO. The homogeneity of elemental distribution suggests that the two-unit cells seen by high-resolution PXRD are compositionally very similar and may be present as intergrowths.
 | ||
| Fig. 2 STEM-ADF images of (a)–(c) as-made LMNbO, and (d)–(e) Mn and Nb EDS elemental maps. | ||
Magnetic susceptibility shows no evidence of long-range magnetic order down to 5 K. A fit to the Curie Weiss law at T > 150 K gave a Curie constant consistent with Mn2+ ions and a Weiss temperature of −159(1) K, suggesting antiferromagnetic interactions, but with structural disorder or magnetic frustration giving only short-range correlations between neighbouring Mn ions, similar to as previously reported in Mn3+ DRS lithium niobate materials.13
LMNbO was ball milled to allow good mixing between active material and carbon black (C65) for electrode preparation. The ball milled sample used in the electrochemical characterisation was found to maintain its crystallinity after this treatment, (Fig. S9, ESI†). SEM images of as-made and ball milled LMNbO showed similar particle size between as-made and milled active material with C65, Fig. S10 (ESI†). Initial electrochemical results using a standard electrolyte (1.0 M LiPF6 in ethylene carbonate (EC)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl methyl carbonate (EMC) in a 1:1 ratio with 1% vinylene carbonate (VC)) demonstrated that the cathode has a high operating voltage evidenced by its plateau above 4.6 V, Fig. 3a. A voltage screening between 4.3 and 4.8 V to 1.5 V determined the correct voltage range to control the redox processes.
:ethyl methyl carbonate (EMC) in a 1:1 ratio with 1% vinylene carbonate (VC)) demonstrated that the cathode has a high operating voltage evidenced by its plateau above 4.6 V, Fig. 3a. A voltage screening between 4.3 and 4.8 V to 1.5 V determined the correct voltage range to control the redox processes.
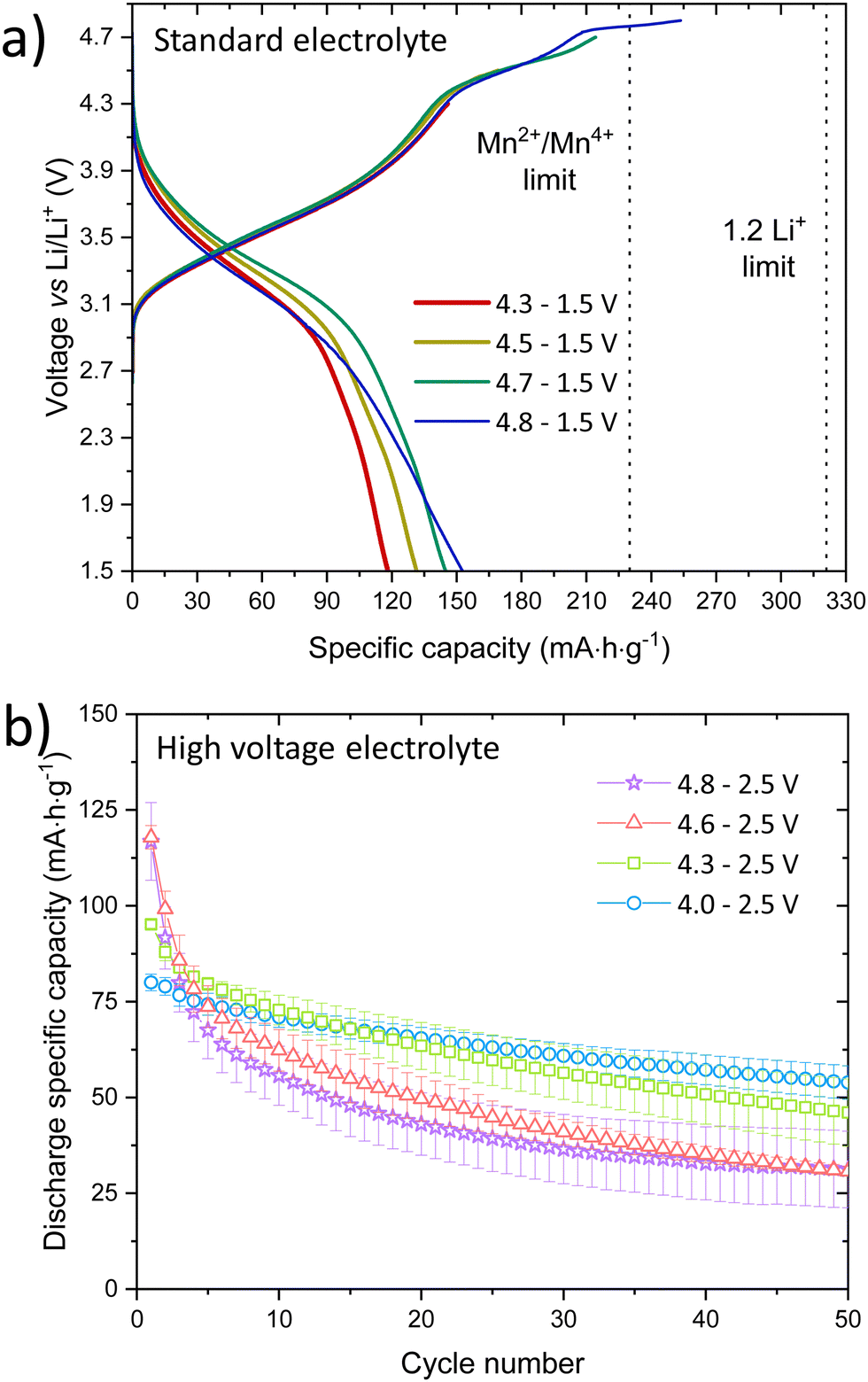 | ||
| Fig. 3 (a) First charge/discharge curves in a standard electrolyte and (b) comparison of discharged capacities over different voltage ranges, where 4.0–2.5 V showed less capacity fading in a high voltage electrolyte. | ||
To minimise Mn dissolution and inhibit the capacity fading due to electrolyte decomposition at high voltages, cycling was performed using a high voltage electrolyte (1 M LiPF6 in 30:70 EC:EMC 2 wt% VC mixed with 2 wt% prop-1-ene-1,3 sultone (PES)), 1wt% methylene methanedisulfonate (MMDS) and 1 wt% tris(trimethylsilyl) phosphite (TTSPi). Table S4 (ESI†) summarises the cycling measurements at 5 mA g−1 over 50 cycles in 4 different voltage windows (up to 4.0, 4.3, 4.6, 4.8 V). Increasing the upper voltage limit, increases the voltage hysteresis. Up to 4.3 V, no voltage plateau is observed and only Mn oxidation occurs. On the other hand, above 4.6 V most of the expected delithiation capacity is obtained, 355 mA h g−1. Compared to the oxyfluoride Mn2+-DRS materials previously reported from solid state and mechanochemical synthesis, LMNbO has one of the highest charge capacities in the first cycle at 4.8 V. LMNbO has a theoretical capacity of 230 mA h g−1 based on Mn oxidation only (Mn2+ to Mn4+), but complete Li extraction would give a theoretical capacity of 321 mA h g−1, if oxygen oxidation is considered.
Even though the first delithiation gives a high capacity, there is a decrease in the overall capacity after 50 cycles from 4.8 to 2.5 V, Fig. 3b. A wider voltage window affects the hysteresis in the first 10 cycles as depicted in Fig. S11 and S12 (ESI†), which decreases when cycled from 4.0 to 2.0 V between the charge and discharge capacities. Electrochemical impedance spectroscopy, Fig. S13 and S14 (ESI†), shows a high charge-transfer resistivity in the first cycle due to overpotential, but a diffusion-controlled process is observed from 4.5 V at low frequencies, indicating diffusion of redox species at the electrode surface.
Ex situ PXRD, Mn K-edge XANES and XPS were used to investigate the capacity fade and the structural stability of the active material when discharged to 1.5 V in a standard electrolyte. PXRD data are shown in Fig. 4, and Pawley refinement from the ex situ PXRD from pristine and delithiated/lithiated electrodes reveals that during delithiation the unit cell decreases in size as Li is extracted and Mn is oxidised, Table S6 (ESI†). On the other hand, when lithiated to 1.5 V, the structure converts into two different rock-salt structures with a = 4.25 and 4.20 Å, which differs from the as-made sample (a = 4.26 and 4.24 Å) and pristine electrode (a = 4.23 Å), suggesting structural instability.
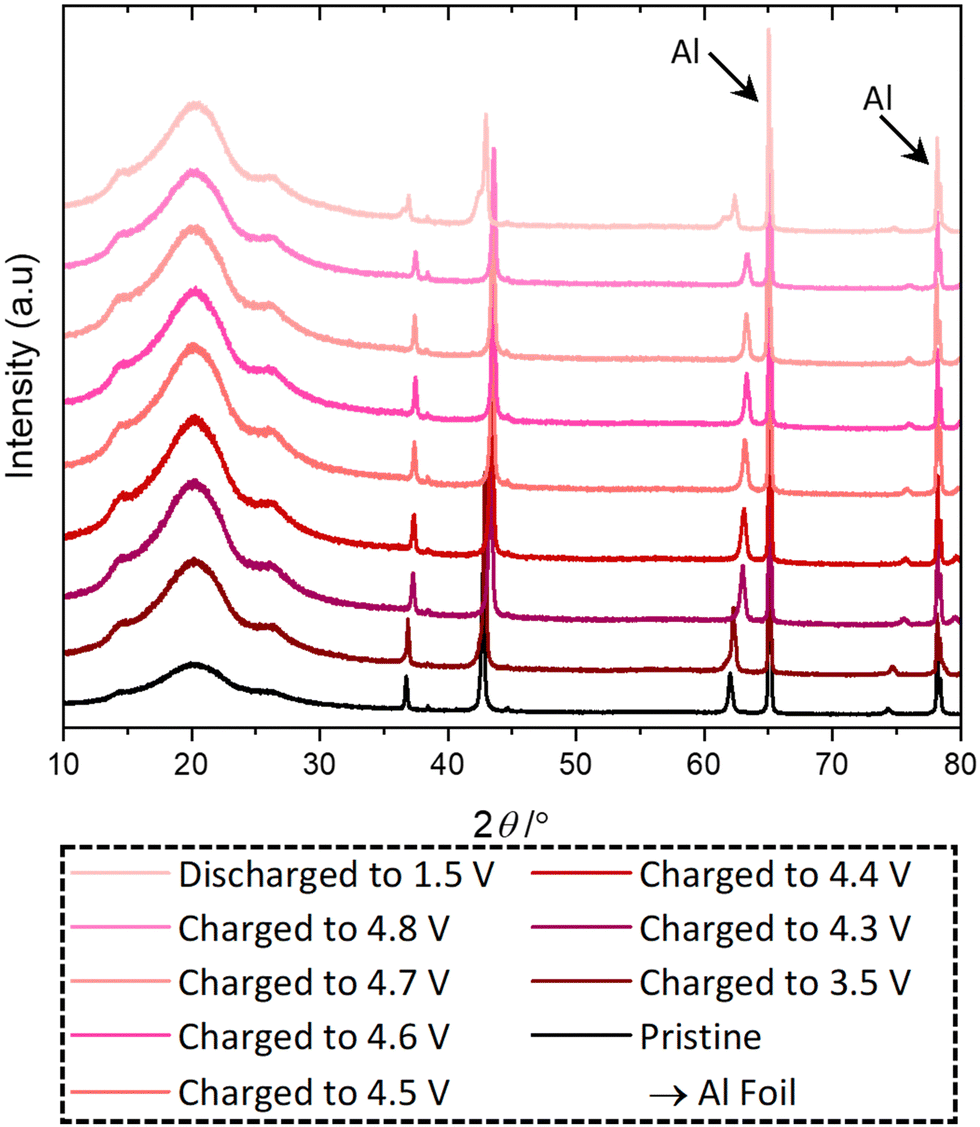 | ||
| Fig. 4 Ex situ PXRD of pristine and cycled LMNbO electrodes. | ||
Ex situ Mn K-edge spectra demonstrate that the pristine electrode has an oxidation state near Mn2+ as expected, Fig. 5. When charged to 3.5 V only a small increase in oxidation state is observed. However, at 4.6 V, most of Mn2+ is oxidised, Mn4+, which is then reduced at 4.8 V to an average oxidation state of approximately 3.3+. This suggests that O2− or other species play a fundamental role to reduce Mn at high voltages and this in turn may contribute to the ultimate structural instability. This is consistent with other studies on the oxygen redox and loss in DRS materials14–16 and Li niobates.17 When Li ions are reinserted, Mn does not return to its original oxidation state, which is the origin of the voltage hysteresis in the first cycle.
 | ||
| Fig. 5 Mn K-edge spectra of (a) pristine and charged electrodes at 3.5, 4.6, 4.7, 4.8 V, and discharged electrode at 1.5 V, and (b) linear fit and oxidation state of Mn in different voltage points. | ||
The dQ/dV plots for the upper voltages of 4.3, 4.5, 4.7 and 4.8, Fig. S18 (ESI†), depict a significant and intense oxidation process above 4.7 V, and summarises the Mn oxidation state at 1.5, 3.5, 4.6, 4.7 and 4.8 V. Nb is electrochemically inactive as previously reported for other Nb-based DRS cathodes18,19 and evidenced in the unchanged Nb XPS spectra from as-made, ball-milled and electrodes, and the voltage profile when tested from 1.5 or 2.5 V to 4.8 V.
Due to their high reactivity, surface oxygen species produced from redox processes are likely to react with the electrolyte. This is supported by the increase in relative intensity of carbonates signal in the XPS as samples are subjected to higher voltage. This is consistent with the idea of oxygen loss in LMNbO and has been seen in other materials such as Li5FeO4,20 LixNi0.13Co0.13Mn0.54O2−δ,21 Li1.3−xNb0.3Fe0.4O2−δ,22 and Li1.2Ni0.2Mn0.6O2.14
In summary, we have demonstrated the synthesis of a Mn2+-Nb-Li disordered rock-salt material using reducing conditions to stabilise the desired Mn oxidation state. To our knowledge, this is the first example of a disordered lithium niobate rock-salt that contains solely Mn2+ without fluorination to stabilise this low oxidation state. Electrochemically, when used as a positive electrode a high voltage plateau is observed. A large hysteresis is observed in the first cycle in the range of 4.8–1.5 V due to oxygen loss reducing Mn, which is supported by ex situ Mn K-edge data. This likely to contribute to structural instability shown in the ex situ PXRD data. The synthetic methodology opens new avenues to explore low oxidation states of transition metals in DRS materials and provides a new approach to synthesise high voltage cathode materials for the next generation of cathode materials in lithium-ion batteries. Further work is required to reduce particle size and resistivity to improve electrochemical performance, and elemental substitution and solid-solutions with other transition metals would be useful to study to explore a new family of materials.
We thank Companhia Brasileira de Metalurgia e Mineração (CBMM) for funding and the University of Warwick's Research Technology Platforms for provision of some of the instrumentation used. M. W. acknowledges financial support from the Engineering and Physical Sciences Research Council (EPSRC)-funded Warwick Analytical Science Centre (EP/V007688/1). Access to I11 at Diamond Light Source was provided by the Block Allocation Group award “Oxford/Warwick Solid State Chemistry BAG to probe composition-structure-property relationships in solids” (CY25166) and access to B18 was provided by the Energy Materials Block Allocation Group SP14239. We are grateful to Dr Giannantonio Cibin for their help with data acquisition on B18. A. S. M would like to acknowledge the Faraday Institution FutureCat (FIRG017) project.
Conflicts of interest
There are no conflicts to declare.References
- J. Ahn, D. Chen and G. Chen, Adv. Energy Mater., 2020, 10, 2001671 CrossRef CAS.
- D. Chen, J. Ahn, E. Self, J. Nanda and G. Chen, J. Mater. Chem. A, 2021, 9, 7826–7837 RSC.
- J. Lee, D. A. Kitchaev, D. H. Kwon, C. W. Lee, J. K. Papp, Y. S. Liu, Z. Y. Lun, R. J. Clement, T. Shi, B. D. McCloskey, J. H. Guo, M. Balasubramanian and G. Ceder, Nature, 2018, 556, 185–190 CrossRef CAS PubMed.
- H. Li, R. Fong, M. Woo, H. Ahmed, D.-H. Seo, R. Malik and J. Lee, Joule, 2022, 6, 53–91 CrossRef CAS.
- D. Chen, J. Ahn and G. Chen, ACS Energy Lett., 2021, 6, 1358–1376 CrossRef CAS.
- R. J. Clement, Z. Lun and G. Ceder, Energy Environ. Sci., 2020, 13, 345–373 RSC.
- M. Nakajima and N. Yabuuchi, Chem. Mater., 2017, 29, 6927–6935 CrossRef CAS.
- G.-H. Lee, J. Lim, J. Shin, L. J. J. Hardwick and W. Yang, Front. Chem., 2023, 11, 1098460 CrossRef CAS PubMed.
- M. Menetrier, J. Bains, L. Croguennec, A. Flambard, E. Bekaert, C. Jordy, P. Biensan and C. Delmas, J. Solid State Chem., 2008, 181, 3303–3307 CrossRef CAS.
- H. Chung, Z. Lebens-Higgins, B. Sayahpour, C. Mejia, A. Grenier, G. E. Kamm, Y. Li, R. Huang, L. F. J. Piper, K. W. Chapman, J.-M. Doux and Y. S. Meng, J. Mater. Chem. A, 2021, 9, 1720–1732 RSC.
- D. R. Modeshia, R. I. Walton, M. R. Mitchell and S. E. Ashbrook, Dalton Trans., 2010, 39, 6031–6036 RSC.
- R. D. Shannon and C. T. Prewitt, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 925–946 CrossRef CAS.
- M. A. Jones, P. J. Reeves, I. D. Seymour, M. J. Cliffe, S. E. Dutton and C. P. Grey, Chem. Commun., 2019, 55, 9027–9030 RSC.
- A. J. Naylor, E. Makkos, J. Maibach, N. Guerrini, A. Sobkowiak, E. Bjoerklund, J. G. Lozano, A. S. Menon, R. Younesi, M. R. Roberts, K. Edstroem, M. S. Islam and P. G. Bruce, J. Mater. Chem. A, 2019, 7, 25355–25368 RSC.
- Y. Zhang, M. M. Wu, J. W. Ma, G. F. Wei, Y. Ling, R. Y. Zhang and Y. H. Huang, ACS Cent. Sci., 2020, 6, 232–240 CrossRef CAS PubMed.
- Y. C. Lu, E. J. Crumlin, G. M. Veith, J. R. Harding, E. Mutoro, L. Baggetto, N. J. Dudney, Z. Liu and Y. Shao-Horn, Sci. Rep., 2012, 2, 715 CrossRef PubMed.
- W. H. Kan, D. Chen, J. K. Papp, A. K. Shukla, A. Huq, C. M. Brown, B. D. McCloskey and G. Chen, Chem. Mater., 2018, 30, 1655–1666 CrossRef CAS.
- N. Yabuuchi, M. Takeuchi, M. Nakayama, H. Shiiba, M. Ogawa, K. Nakayama, T. Ohta, D. Endo, T. Ozaki, T. Inamasu, K. Sato and S. Komaba, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7650–7655 CrossRef CAS PubMed.
- Z. Lun, B. Ouyang, D.-H. Kwon, Y. Ha, E. E. Foley, T.-Y. Huang, Z. Cai, H. Kim, M. Balasubramanian, Y. Sun, J. Huang, Y. Tian, H. Kim, B. D. McCloskey, W. Yang, R. J. Clement, H. Ji and G. Ceder, Nat. Mater., 2021, 20, 214–221 CrossRef CAS PubMed.
- C. Zhan, Z. P. Yao, J. Lu, L. Ma, V. A. Maroni, L. Li, E. Lee, E. E. Alp, T. P. Wu, J. G. Wen, Y. Ren, C. Johnson, M. M. Thackeray, M. K. Y. Chan, C. Wolverton and K. Amine, Nat. Energy, 2017, 2, 963–971 CrossRef CAS.
- N. Yabuuchi, K. Yoshii, S. T. Myung, I. Nakai and S. Komaba, J. Am. Chem. Soc., 2011, 133, 4404–4419 CrossRef CAS PubMed.
- N. Yabuuchi, M. Nakayama, M. Takeuchi, S. Komaba, Y. Hashimoto, T. Mukai, H. Shiiba, K. Sato, Y. Kobayashi, A. Nakao, M. Yonemura, K. Yamanaka, K. Mitsuhara and T. Ohta, Nat. Commun., 2016, 7, 13814 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc04264b |
| This journal is © The Royal Society of Chemistry 2023 |