
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoswitchable electron-rich phosphines: using light to modulate the electron-donating ability of phosphines†‡
Florenz
Buß
a,
Mowpriya
Das
b,
Daniel
Janssen-Müller
bc,
Alexander
Sietmann
d,
Ankita
Das
b,
Lukas F. B.
Wilm
a,
Matthias
Freitag
b,
Michael
Seidl
d,
Frank
Glorius
 *b and
Fabian
Dielmann
*ad
*b and
Fabian
Dielmann
*ad
aWestfälische Wilhelms-Universität Münster, Institut für Anorganische und Analytische Chemie, Corrensstraße 30, Münster 48149, Germany
bWestfälische Wilhelms-Universität Münster, Organisch-Chemisches Institut, Corrensstraße 36, Münster 48149, Germany. E-mail: glorius@uni-muenster.de
cInstitut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen, Tammannstr. 2, Göttingen 37077, Germany
dInstitute of General, Inorganic and Theoretical Chemistry, University of Innsbruck, Center for Chemistry and Biomedicine, Innrain 80-82, Innsbruck A-6020, Austria. E-mail: fabian.dielmann@uibk.ac.at
First published on 12th September 2023
Abstract
The synthesis and properties of photoswitchable electron-rich phosphines containing N-heterocyclic imines equipped with a photochromic dithienylethene unit are reported. Heteronuclear NMR spectroscopy and UV/vis studies reveal that the imine substituents undergo reversible electrocyclic ring-closing and ring-opening reactions upon exposure to UV and visible light, respectively. The photoisomerization alters the electron-donating ability of the phosphines by up to ΔTEP = 8 cm−1.
Advances in modern transition metal and main group chemistry have been driven by the availability of strong donor ligands such as tertiary phosphines or N-heterocyclic carbenes (NHCs) with tuneable steric and electronic properties.1–6 Using light to modulate the electron-donating character of these ligands is a particularly intriguing approach to remotely control properties of coordination compounds and the outcome of chemical reactions.7–14 Dithienylethene (DTE) compounds, which were first introduced by Irie and co-workers,15–18 have been proven most effective to alter the electron-donating ability of these ligands. Several photoresponsive phosphines with diazobenzene19–24 and stiff-stilbene25–27 chromophors have been reported. However, the photochemical E/Z isomerization mainly affects their steric properties and has negligible impact on their electronic properties.28 Branda and co-workers reported the first photoswitchable DTE-based phosphine by attaching diphenylphosphino groups to the thiophene rings of the chromophore (Chart 1).29,30 Using the 1JPSe coupling constant to probe the basicity,31 they showed that the diphosphine is significantly more electron rich in the open form (1JPSe = 744 Hz) than in the closed form (1JPSe = 756 Hz), which corresponds to a change in basicity of ΔpKa = 1.6 units. Subsequent studies showed that the variation of the phosphine basicity is less pronounced for the non-fluorinated DTE backbone.32 The first chelating diphosphine which carries the phosphino groups at the central ethene unit of the chromophore was recently reported by Wolf and co-workers (Chart 1). The hydroboration of a styrene derivative is catalyzed by the corresponding phosphine-copper(I) complex at different rates, depending on the closed/open form of the DTE backbone.33 The group of Yam incorporated the DTE unit into a phosphole and showed that the corresponding alkynylgold(I) complexes display photochromic and mechanochromic properties.34
 | ||
| Chart 1 Examples of photoswitchable DTE-based phosphines. | ||
A successful approach to photoswitchable NHCs was reported by the groups of Yam and Bielawski based on the incorporation of a photochromic DTE unit into the NHC-backbone.35–39 The NHC undergoes electrocyclic ring closure upon exposure to UV light leading to a significantly reduced donor strength as determined by the change of the Tolman electronic parameter (TEP)54 from 2049 cm−1 (open form) to 2055 cm−1 (closed form).
The synthetic utilization of these NHCs was demonstrated by enabling photoswitchable Rh(I) catalyzed hydroborations,40 transesterifications, amidations,41 and the reversible activation of ammonia.42 Recently, we converted Bielawski's NHC into N-heterocyclic imines (NHIs) which gave rise to photoswitchable nitrogen superbases exhibiting substantial pKa shifts of up to 8.7 units enabling light-driven reversible CO2 fixation.43 In 2015, we established that the electron-donating power of phosphines is considerably enhanced by substituents with strong π-donor ability such as NHI groups.44 Changing the π-donor ability of the substituents thus directly affects the phosphine donor strength, which becomes particularly evident upon protonation of the exocyclic nitrogen atom of the NHI π system, changing the TEP of the phosphine by about ΔTEP = 20 cm−1 for each protonated substituent.45–47 Small structural variations of the N-heterocyclic π-donor, such as the use of a benzimidazoline rather than an imidazoline backbone, have also been shown to alter the donor properties and reactivity of the corresponding phosphine.48–52 Expanding on these results, we herein use a DTE-annulated NHI substituent to generate electron-rich phosphines with photoswitchable electronic properties (Chart 1).
For a selection of monosubstituted photoswitchable phosphines (PS-IAPs) with diverse donor abilities, we targeted the synthesis of 3-o, 4-o and 5-o (Scheme 1). Henceforth, the notation o and c refer to the ring-opened and -closed form of the DTE unit in PS-IAPs, respectively. Following the reported synthesis of IAPs,44,46,53 the phosphines were obtained as purple solids in yields of 93–97% by deprotonation of iminium salt 2 with n-butyllithium and subsequent treatment with the respective chlorophosphine. The phosphines, which can be stored in the absence of air and moisture for months, show good solubility in n-hexane, toluene, Et2O, THF and acetonitrile.
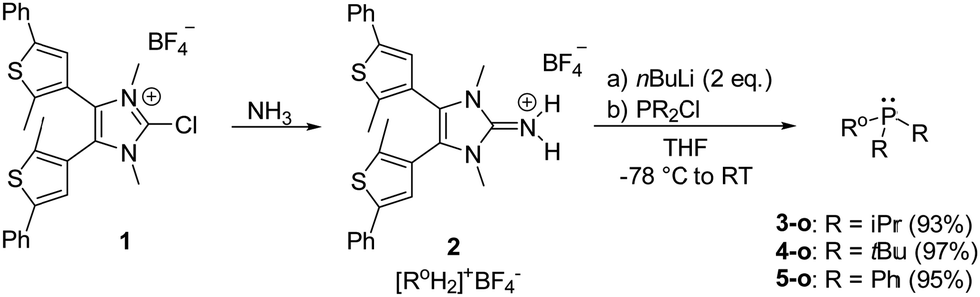 | ||
| Scheme 1 Synthesis of iminium salt 2 and preparation of mono-substituted PS-IAPs 3-o, 4-o, and 5-o. | ||
As the DTE unit in the backbone of 3-o, 4-o and 5-o is expected to undergo a reversible electrocyclic photoisomerization reaction, the UV/vis and NMR spectroscopic properties of the phosphines were studied after exposure to UV and visible light, respectively. The UV/vis spectrum of the colorless solution of 3-o in THF exhibits an intense absorption centered at 290 nm. Subsequent UV irradiation (λirr = 315 nm) resulted in a purple solution and the decrease of the band centered at 290 nm concomitant with the appearance of new bands at 403 and 590 nm (Fig. 1). The initial UV/vis spectrum can be restored by irradiation of the solution at 585 nm for 5 minutes. Similar behavior was observed for compounds 4-o and 5-o (see the ESI†).
 | ||
| Fig. 1 UV/vis spectral changes of 3-o in THF ([3-o]0 = 10−4 M) upon UV irradiation (λirr = 315 nm, 140 mW). The spectra were recorded after irradiation for 0 min (green line) and 3 min (blue line). The arrows indicate the evolution of the spectral changes. | ||
31P NMR spectroscopy is a convenient tool to study small changes in the electronic properties of guanidine-substituted phosphines,44,46,53 and thus to monitor the photocyclization reaction of PS-IAPs. In fact, upon irradiation of a THF-d8 solution of 3-o at 310 nm (140 mW) for 1 hour in a quartz NMR tube, a new signal appeared at 58.5 ppm in the 31P NMR spectrum, which is shifted upfield compared to that of 3-o (59.6 ppm) and corresponds to the ring-closed species 3-c. In the 1H NMR spectrum (in THF-d8), the two diagnostic resonances of the thiophen protons shift from 6.87 to 6.91 ppm and the methyl groups at the imidazole unit from 3.36 to 3.61 ppm.
Quantitative 31P and 1H NMR spectroscopy reveal 86% conversion of 3-o to 3-c upon irradiation with UV light (λirr = 315 nm, 140 mW) for 1 hour. Prolonged irradiation of the mixture by up to 12 h did not increase this value, which can be attributed to the superimposed UV/vis absorption bands of 3-o and 3-c (Fig. 1). The solution containing 3-o and 3-c can be stored in the dark for one week without noticeable changes in the 31P and 1H NMR spectra. However, full conversion back to 3-o upon irradiation with visible light (λirr = 585 nm, 140 mW) for one hour was accomplished. Similar spectral changes were observed for compounds 4-o and 5-o upon UV/vis-initiated ring-closing and ring-opening showing 88% and 90% photoconversion to the ring-closed form, respectively, and complete conversion to the ring-open form. The 31P NMR resonances shift from 72.8 ppm (4-o) to 71.5 ppm (5-c) and from 33.8 ppm (5-o) to 31.9 ppm (5-c). For fatigue resistance testing, a concentrated solution of 5-o was irradiated cyclically with UV and visible radiation for 1 and 3 hours, respectively. After 10 cycles, 31P NMR analysis of the solution reveals about 14% degradation of the phosphine.
The effect of photoisomerization on the donor strength of the new phosphines was examined by determining their TEP values via IR spectroscopic characterization of the corresponding nickel complexes [(PS-IAP)Ni(CO)3] (Scheme 2).§ The electrocyclic ring closure of the NHI backbone upon UV irradiation leads to significantly reduced donor strength of the corresponding phosphines. The change in TEP values due to photocyclization of a single NHI group (ΔTEP of 7–8 cm−1) exceeds that of photochromic NHCs (ΔTEP = 6 cm−1).40,41 Taking the TEP values of phosphines 3, 4 and 5 into account, single substituent parameters of χ = −9.5 and χ = −1.9 can be derived for the open and closed state of the photochromic imine substituent, respectively. Similar substituent parameters were observed for NHIs with 4,5-dimethylimidazoline (χ = −8.8 cm−1) and benzimidazoline (χ = −3.9 cm−1) backbones,44,49 showing that the photoisomerization of the DTE unit significantly affects the π-donor character of the NHI substituent.
 | ||
| Scheme 2 Preparation of the nickel complexes [(PS-IAP)Ni(CO)3] and the change of the TEP values upon light-induced photoisomerization of the DTE backbone. | ||
The molecular structure of 7-o was established using single-crystal X-ray crystallography (Fig. 2). The thiophene rings are tilted from the plane of the imidazole ring (56°/42°). Thus, π conjugation from the electron rich NHI unit into the thiophene rings is prevented, which is also evident by the C2–C6 and C3–C17 bond distances in the range of single bonds and explains the strong π-donor character of the substituent in the open state.
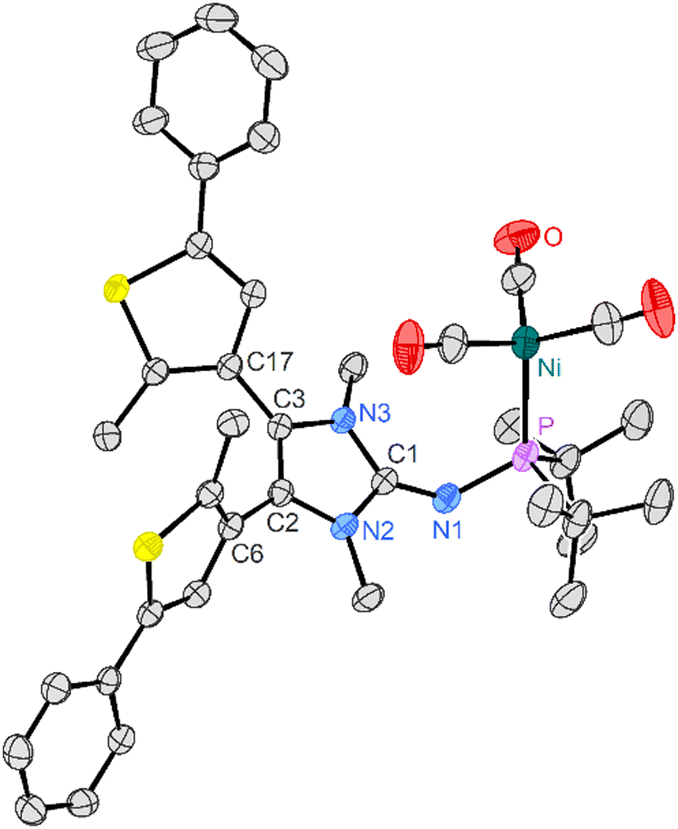 | ||
| Fig. 2 Solid-state structure of 7-o. Hydrogen atoms and a disordered pentane solvent molecule are omitted for clarity. Ellipsoids are drawn at 50% probability. Selected bond lengths [Å]: P–Ni 2.2717(5), P–N1 1.6624(16), N1–C1 1.306(2), N2–C1 1.376(2), N3–C1 1.378(2), N2–C2 1.395(2), N3–C3 1.405(2), C2–C3 1.357(2), C2–C6 1.468(2), C3–C17 1.464(2). | ||
We previously demonstrated that the individual contribution of each π donor substituent to the overall donor ability of IAPs are additive,44,51 in agreement with Tolman's additivity rule.54 A phosphine with three photoswitchable NHI substituents is therefore predicted to undergo photoinduced TEP changes of up to 21.6 cm−1 in the range of 2027.0 cm−1 (10-o) to 2048.6 cm−1 (10-c). We attempted the synthesis of 10-o by in situ deprotonation of 2 with nBuLi or KHMDS and reaction with PCl3. However, an insoluble solid material was obtained, presumably due to the formation of stable coordination compounds via coordination of the sterically accessible exocyclic N atoms of the NHI moieties to alkali metal salts. Phosphonium salt [10-o]HCl was therefore prepared in an alkali metal-free route from the reaction of imine 9 with P(NMe2)2Cl (Scheme 3). [10-o]HCl was isolated as a white solid in 59% yield. However, treatment of [10-o]HCl with inorganic bases led to the same solubility issues. Using the nonionic t-Bu-P4 Schwesinger base, the deprotonation of [10-o]HCl was successful. In the 31P NMR spectrum of the reaction mixture, the signal for [10-o]HCl (−22.3 ppm) had disappeared and a new resonance appeared at 75.9 ppm with the expected (P(NIiPr)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :79.6 ppm)49 chemical shift of the desired PS-IAP 10-o. Despite several attempts, PS-IAP 10-o could not be separated from [t-Bu-P4]·HCl via extraction with n-hexane, n-pentane, diethyl ether or benzene, indicating that 10-o is sparingly soluble in these solvents. Similar to the PS-IAPs with one NHI substituents, the UV/vis spectrum of the THF solution containing 10-o and [t-Bu-P4]·HCl in THF exhibits an absorption centered at 290 nm. Exposure of the solution to UV light (λirr = 310 nm, 30 s) resulted in a deep purple solution and the decrease of the band centered at 290 nm concomitant with the appearance of new bands at 385 and 650 nm (Fig. S57, ESI†). These bands decrease upon prolongated irradiation and new absorption bands appear at 355 nm and 580 nm. This observation suggests the stepwise photocyclization of the NHI substituents leading to several isomers. Indeed, the initial UV/vis spectra can be restored by exposure to light at 585 nm for 5 minutes, confirming the reversibility of the photoreaction.
:79.6 ppm)49 chemical shift of the desired PS-IAP 10-o. Despite several attempts, PS-IAP 10-o could not be separated from [t-Bu-P4]·HCl via extraction with n-hexane, n-pentane, diethyl ether or benzene, indicating that 10-o is sparingly soluble in these solvents. Similar to the PS-IAPs with one NHI substituents, the UV/vis spectrum of the THF solution containing 10-o and [t-Bu-P4]·HCl in THF exhibits an absorption centered at 290 nm. Exposure of the solution to UV light (λirr = 310 nm, 30 s) resulted in a deep purple solution and the decrease of the band centered at 290 nm concomitant with the appearance of new bands at 385 and 650 nm (Fig. S57, ESI†). These bands decrease upon prolongated irradiation and new absorption bands appear at 355 nm and 580 nm. This observation suggests the stepwise photocyclization of the NHI substituents leading to several isomers. Indeed, the initial UV/vis spectra can be restored by exposure to light at 585 nm for 5 minutes, confirming the reversibility of the photoreaction.
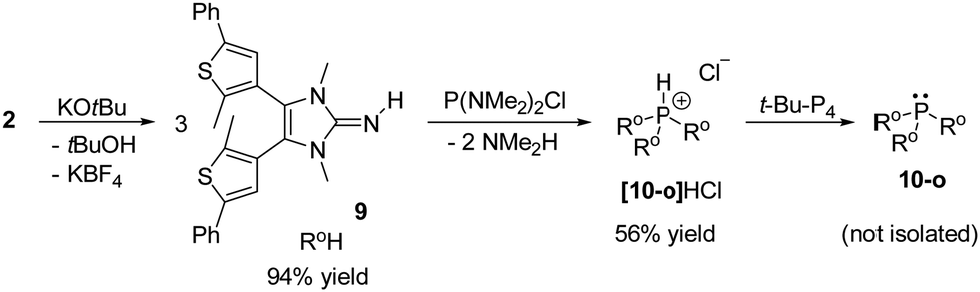 | ||
| Scheme 3 Synthesis of the protonated tri-substituted PS-IAP [10-o]HCl and generation of the free phosphine 10-o by in situ deprotonation. | ||
In conclusion, three photoswitchable phosphines (PS-IAPs) with electron donor ability in the range of NHCs were synthesized. Upon irradiation with UV and visible light, the photochromic DTE unit undergoes reversible photocyclization reaction with 85% conversion leading to changes in the TEP values of the corresponding phosphines of up to ΔTEP = 8 cm−1, which exceeds that of photochromic NHCs (ΔTEP = 6 cm−1).40,41 Photo-induced TEP changes of up to ΔTEP = 21.6 cm−1 are predicted for PS-IAPs with more than one photoswitchable NHI substituent. However, issues with the isolation of 10-o indicate that sterically more encumbered substituents are recommended to prevent the formation of stable coordination compounds and to increase the solubility of the PS-IAPs.
D. J.-M., M. D., M. F. and A. D. synthesized imidazolium salt 1. F. B. and A. S. synthesized the phosphines, supported by M. D. and L. F. B. W, and performed the photochemical experiments. M. S. performed the SCXRD study. F. D. and F. G. directed the investigation. F. B. and F. D. wrote the manuscript with contributions from all authors. All authors have given approval to the final version of the manuscript.
The authors gratefully acknowledge financial support from the DFG (Emmy Noether program: DI2054/1-1, SFB 858, IRTG 2027). Thanks are due to the German Academic Scholarship Foundation for a PhD fellowship (L.W.). We thank Dr Marius Wünsche for providing suggestions at different stages of this study.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef PubMed.
- J. A. Gillespie, E. Zuidema, P. W. N. M. van Leeuwen and P. C. J. Kamer, in Phosphorus (III) ligands in homogeneous catalysis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, Wiley-Blackwell, Oxford, 2012, pp. 1–26 Search PubMed.
- V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed.
- A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed.
- P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem., 2021, 5, 711–725 CrossRef CAS PubMed.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- Z. Freixa, Catal. Sci. Technol., 2020, 10, 3122–3139 RSC.
- D. Majee and S. Presolski, ACS Catal., 2021, 11, 2244–2252 CrossRef CAS.
- A. Ghorbani-Choghamarani and Z. Taherinia, RSC Adv., 2022, 12, 23595–23617 RSC.
- B. M. Neilson and C. W. Bielawski, ACS Catal., 2013, 3, 1874–1885 CrossRef CAS.
- R. Göstl, A. Senf and S. Hecht, Chem. Soc. Rev., 2014, 43, 1982–1996 RSC.
- D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef PubMed.
- V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341–5370 RSC.
- R. Dorel and B. L. Feringa, Chem. Commun., 2019, 55, 6477–6486 RSC.
- R. M. Kellogg, M. B. Groen and H. Wynberg, J. Org. Chem., 1967, 32, 3093–3100 CrossRef CAS.
- M. Irie and M. Mohri, J. Org. Chem., 1988, 53, 803–808 CrossRef CAS.
- L. N. Lucas, J. van Esch, R. M. Kellogg and B. L. Feringa, Chem. Commun., 1998, 2313–2314 RSC.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- M. Kawamura, R. Kiyotake and K. Kudo, Chirality, 2002, 14, 724–726 CrossRef CAS PubMed.
- M. Yamamura, N. Kano and T. Kawashima, J. Am. Chem. Soc., 2005, 127, 11954–11955 CrossRef CAS PubMed.
- M. D. Segarra-Maset, P. W. N. M. van Leeuwen and Z. Freixa, Eur. J. Inorg. Chem., 2010, 2075–2078 CrossRef CAS.
- H. Bricout, E. Banaszak, C. Len, F. Hapiot and E. Monflier, Chem. Commun., 2010, 46, 7813–7815 RSC.
- N. Priyadarshani, B. Ginovska, J. T. Bays, J. C. Linehan and W. J. Shaw, Dalton Trans., 2015, 44, 14854–14864 RSC.
- T. Arif, C. Cazorla, N. Bogliotti, N. Saleh, F. Blanchard, V. Gandon, R. Métivier, J. Xie, A. Voituriez and A. Marinetti, Catal. Sci. Technol., 2018, 8, 710–715 RSC.
- Z. S. Kean, S. Akbulatov, Y. Tian, R. A. Widenhoefer, R. Boulatov and S. L. Craig, Angew. Chem., Int. Ed., 2014, 53, 14508–14511 CrossRef CAS PubMed.
- D. Zhao, T. M. Neubauer and B. L. Feringa, Nat. Commun., 2015, 6, 6652 CrossRef CAS PubMed.
- R. Costil, S. Crespi, L. Pfeifer and B. L. Feringa, Chem. – Eur. J., 2020, 26, 7783–7787 CrossRef CAS PubMed.
- F. Medici, N. Goual, V. Delattre, A. Voituriez and A. Marinetti, ChemCatChem, 2020, 12, 5573–5589 CrossRef CAS.
- D. Sud, R. McDonald and N. R. Branda, Inorg. Chem., 2005, 44, 5960–5962 CrossRef CAS PubMed.
- H. D. Samachetty and N. R. Branda, Pure Appl. Chem., 2006, 78, 2351–2359 CrossRef CAS.
- U. Beckmann, D. Süslüyan and P. C. Kunz, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 2061–2070 CrossRef CAS.
- G. Bianchini, G. Strukul, D. F. Wass and A. Scarso, RSC Adv., 2015, 5, 10795–10798 RSC.
- Z. Xu, Y. Cao, B. O. Patrick and M. O. Wolf, Chem. – Eur. J., 2018, 24, 10315–10319 CrossRef CAS PubMed.
- N. M.-W. Wu, M. Ng and V. W.-W. Yam, Angew. Chem., Int. Ed., 2019, 58, 3027–3031 CrossRef CAS PubMed.
- T. Nakashima, M. Goto, S. Kawai and T. Kawai, J. Am. Chem. Soc., 2008, 130, 14570–14575 CrossRef CAS PubMed.
- V. W.-W. Yam, J. K.-W. Lee, C.-C. Ko and N. Zhu, J. Am. Chem. Soc., 2009, 131, 912–913 CrossRef CAS PubMed.
- G. Duan, N. Zhu and V. W.-W. Yam, Chem. – Eur. J., 2010, 16, 13199–13209 CrossRef CAS PubMed.
- B. M. Neilson, V. M. Lynch and C. W. Bielawski, Angew. Chem., Int. Ed., 2011, 50, 10322–10326 CrossRef CAS PubMed.
- G. Duan, W.-T. Wong and V. W.-W. Yam, New J. Chem., 2011, 35, 2267 RSC.
- B. M. Neilson and C. W. Bielawski, Organometallics, 2013, 32, 3121–3128 CrossRef CAS.
- B. M. Neilson and C. W. Bielawski, J. Am. Chem. Soc., 2012, 134, 12693–12699 CrossRef CAS PubMed.
- A. J. Teator, Y. Tian, M. Chen, J. K. Lee and C. W. Bielawski, Angew. Chem., Int. Ed., 2015, 54, 11559–11563 CrossRef CAS PubMed.
- L. F. B. Wilm, M. Das, D. Janssen-Müller, C. Mück-Lichtenfeld, F. Glorius and F. Dielmann, Angew. Chem., Int. Ed., 2022, 61, e202112344 CrossRef CAS PubMed.
- M. A. Wünsche, P. Mehlmann, T. Witteler, F. Buß, P. Rathmann and F. Dielmann, Angew. Chem., Int. Ed., 2015, 54, 11857–11860 CrossRef PubMed.
- P. Mehlmann and F. Dielmann, Chem. – Eur. J., 2019, 25, 2352–2357 CrossRef CAS PubMed.
- J. A. Werra, M. A. Wünsche, P. Rathmann, P. Mehlmann, P. Löwe and F. Dielmann, Z. Anorg. Allg. Chem., 2020, 646, 794–799 CrossRef CAS.
- B. S. Birenheide, F. Krämer, L. Bayer, P. Mehlmann, F. Dielmann and F. Breher, Chem. – Eur. J., 2021, 27, 15066–15073 CAS.
- F. Buß, P. Mehlmann, C. Mück-Lichtenfeld, K. Bergander and F. Dielmann, J. Am. Chem. Soc., 2016, 138, 1840–1843 CrossRef PubMed.
- P. Mehlmann, C. Mück-Lichtenfeld, T. T. Y. Tan and F. Dielmann, Chem. – Eur. J., 2017, 23, 5929–5933 CrossRef CAS PubMed.
- T. Witteler, H. Darmandeh, P. Mehlmann and F. Dielmann, Organometallics, 2018, 37, 3064–3072 CrossRef CAS.
- F. Buß, P. Rotering, C. Mück-Lichtenfeld and F. Dielmann, Dalton Trans., 2018, 47, 10420–10424 RSC.
- F. Buß, C. Mück-Lichtenfeld, P. Mehlmann and F. Dielmann, Angew. Chem., Int. Ed., 2018, 57, 4951–4955 CrossRef PubMed.
- P. Rotering, L. F. B. Wilm, J. A. Werra and F. Dielmann, Chem. – Eur. J., 2020, 26, 406–411 CrossRef CAS PubMed.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
Footnotes |
| † Dedicated to Holger Braunschweig on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2289253. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc04050j |
| § Note that complex 8-o did not undergo the electrocyclic ring-closing reaction upon UV irradiation, presumably due to overlapping absorption bands. The ring-closed phosphine 5-o was therefore reacted with Ni(CO)4 to access 8-c. |
| This journal is © The Royal Society of Chemistry 2023 |