
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly selective CO2 electrolysis in aqueous media by a water-soluble cobalt dimethyl-bipyridine complex†
Tomiko M.
Suzuki
 *a,
Kengo
Nagatsuka
b,
Takamasa
Nonaka
a,
Yuichi
Yamaguchi
bc,
Naonari
Sakamoto
a,
Takeshi
Uyama
a,
Keita
Sekizawa
a,
Akihiko
Kudo
*bc and
Takeshi
Morikawa
*a
*a,
Kengo
Nagatsuka
b,
Takamasa
Nonaka
a,
Yuichi
Yamaguchi
bc,
Naonari
Sakamoto
a,
Takeshi
Uyama
a,
Keita
Sekizawa
a,
Akihiko
Kudo
*bc and
Takeshi
Morikawa
*a
aToyota Central R&D Labs., Inc., 41-1 Yokomichi, Nagakute, Aichi 480-1192, Japan. E-mail: tomiko@mosk.tytlabs.co.jp; morikawa@mosk.tytlabs.co.jp
bDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: a-kudo@rs.tus.ac.jp
cCarbon Value Research Center, Research Institute for Science & Technology, Tokyo University of Science, 2641 Yamazaki, Noda-shi, Chiba 278-8510, Japan
First published on 20th September 2023
Abstract
A water-soluble Co complex with dimethyl-bipyridine ligands reduced CO2 to CO electrochemically with almost 100% selectivity at −0.80 V vs. NHE in an aqueous medium (pH 6.8) without an organic solvent. The reaction overpotential was 270 mV. A possible CO formation mechanism was discussed based on experiments and calculations.
Reductive conversion of CO2 into useful energy-rich chemicals using water and sustainable energy is a promising approach to ameliorating the fossil fuel shortage and mitigating global warming. Thus, the electrocatalytic CO2 reduction reaction (CO2RR) to form C1 chemicals such as basic ingredients carbon monoxide (CO) and formic acid is an active research topic. CO is a desirable target product because it can be converted into various hydrocarbons via Fischer–Tropsch synthesis. Gaseous CO exhibits low solubility in aqueous media, making its collection easier than that of liquid products such as formic acid.
Molecular metal complexes are advantageous as catalysts because their properties can be controlled through manipulation of their metal–ligand interactions; some molecular complexes therefore exhibit high reaction selectivity toward CO2, especially in organic solvents with a sacrificial electron donor. The use of water molecules as electron donors, high CO2 selectivity in aqueous solution, and a low overpotential for the CO2RR are all important requirements for a sustainable catalytic system. However, CO2 reduction by complex catalysts dissolved in almost 100% water is challenging. Nonetheless, the literature contains a few reports on selective electrocatalytic or photocatalytic CO2RR by water-soluble metal complexes in a fully aqueous solution. Selective CO2 reduction by water-soluble complex catalysts with macrocyclic ligands such as cyclam, porphyrin, or their derivatives has been reported.1–5 In addition, polypyridines such as 2,2′-bipyridine (bpy), 2,2′:6′,2′′-terpyridine (tpy), and phenanthroline (phen) are common ligands in coordination chemistry and molecular catalysis because they generally construct stable, well-defined complexes. A water-soluble fac-[Re(CH2OH)–OH2]+ (fac-[Re(4,4′-dihydroxymethyl-2,2′-bipyridine)(CO)3(OH2)]+) has been reported to show electrochemical CO production with 95% selectivity at −1.1 V vs. normal hydrogen electrode (NHE) in an aqueous solution at pH 6.9.6 More abundant metal elements are also desired as central metal species that drive an aqueous CO2RR in terms of lowering the cost and reducing CO2 emissions in the overall system lifecycle.7–9 [Mn(bpy(COOH)2)(CO)3Br] (bpy(COOH)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4,4′-dicarboxy-2,2′-bipyridine) and [Cu(phen)2]2+ complexes have also been reported as water-soluble catalysts with a faradaic efficiency (FE) of 65% and ∼60%, respectively, for CO formation for the electrochemical CO2RR (Table S1, ESI†).10,11
:4,4′-dicarboxy-2,2′-bipyridine) and [Cu(phen)2]2+ complexes have also been reported as water-soluble catalysts with a faradaic efficiency (FE) of 65% and ∼60%, respectively, for CO formation for the electrochemical CO2RR (Table S1, ESI†).10,11
Water-soluble Co complexes with bpy ligands have been reported as electro- and photocatalysts for hydrogen evolution,12 whereas their ability to function as a CO2 reduction catalyst in aqueous solution has not yet been demonstrated. In the case of the tpy ligand, [Co(tpy)2]2+ ([Co-tpy]) has been reported to produce CO and H2 electrochemically in a dimethylformamide (DMF)/H2O (95/5 vol%) mixed solvent.13 In the case of the bpy ligand, [Co(bpy)3]2+ ([Co-bpy]) or [Co(dmbpy)3]2+ ([Co-dmbpy], dmbpy: 4,4′-dimethyl-2,2′-bipyridine) has been shown to promote the CO2RR to produce CO and H2 under visible-light irradiation when used in combination with a photosensitizer or a semiconductor photocatalyst in an acetonitrile (MeCN) or MeCN/H2O mixture containing sacrificial electron donor reagents.14–17 We recently adopted a water-soluble [Co-dmbpy] (Fig. 1a) for Z-schematic (two-step photoexcitation) photocatalysis for the CO2RR in an aqueous suspension with two coexisting semiconductors (BiVO4 for water oxidation and (CuGa)0.3Zn1.4S2 for the CO2RR).18 The system demonstrated 98% CO selectivity (against 2% H2) and 1740 μmol-CO was produced using 12 μmol [Co-dmbpy] under visible-light irradiation (wavelength > 420 nm). The [Co-dmbpy] behaved as a redox-shuttle electron mediator for the two coexisting semiconductors. Furthermore, experiments and calculations strongly suggested that the [Co-dmbpy] acted as a highly selective cocatalyst for CO generation by receiving electrons from (CuGa)0.3Zn1.4S2.18 However, the ability of the CO2RR with unusually high selectivity over [Co-dmbpy] in an aqueous solution has not been clarified, even electrochemically.
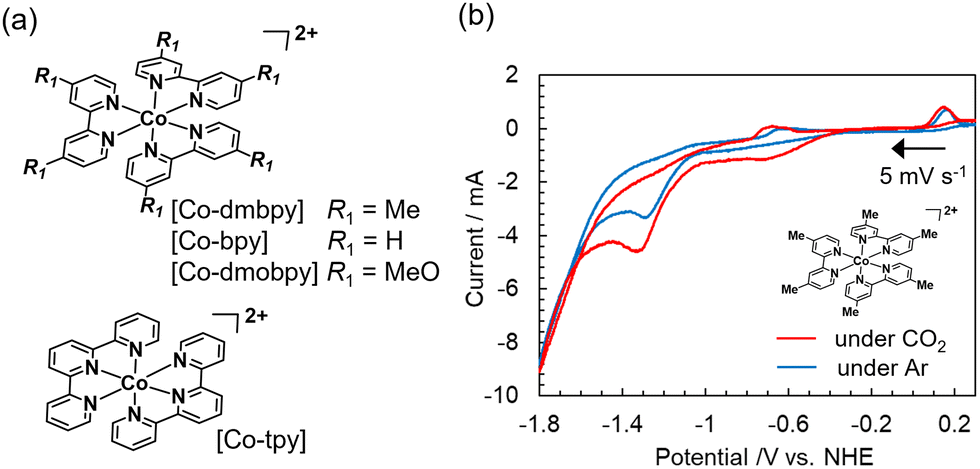 | ||
| Fig. 1 (a) Chemical structure of Co complexes with various polypyridine ligands and NO3− counter anions (dmbpy: 4,4′-dimethyl-2,2′-bipyridine, bpy: 2,2′-bipyridine, dmobpy: 4,4′-dimethoxy-2,2′-bipyridine, tpy: 2,2′:6′,2-terpyridine). (b) CV curves for [Co-dmbpy] (0.3 mmol L−1) in an aqueous NaHCO3 (0.1 mol L−1) solution bubbled with CO2 (red, pH 6.8) or Ar (blue, adjusted to pH 6.8). | ||
In this communication, the electrochemical CO2RR over the water-soluble [Co-dmbpy] was investigated. The experiments clarified that [Co-dmbpy] reduced CO2 to CO with nearly 100% selectivity at a low potential of −0.80 V vs. NHE (pH 6.8, −0.40 V vs. reversible hydrogen electrode (RHE)), and further discussed the CO2RR mechanism in combination with calculation.
Water-soluble divalent Co complexes [Co-R1bpy], which have various substituents R1 at the 2,2′ position of the bpy ligand, and [Co-tpy], which has the tpy ligand (Fig. 1a), were synthesized.18Fig. 1b shows a current–voltage (cyclic voltammogram 〈CV〉) curve for [Co-dmbpy] (0.3 mmol L−1) recorded in an aqueous NaHCO3 (0.1 mol L−1) solution bubbled with CO2 or Ar where carbon paper (CP) and Pt wire were used as working and counter electrodes, respectively. An H-shaped two-compartment reactor separated by a Nafion membrane was used (Fig. S1, ESI†). In the CV curve under a CO2 atmosphere (red line), a small first reduction wave from about −0.4 V vs. NHE (pH 6.8) involving a valence change of the Co ion and a larger second reduction wave from −1.0 V vs. NHE was observed. This second reduction wave is considered as the CO2 reduction current over the complex. On the other hand, the Ar atmosphere (blue line, in the absence of CO2) shows a similar CV curve, but the current values for the two reduction waves are lower than for the CO2 atmosphere, especially the first reduction wave, which yields very small current. We focused on this first reductive peak and performed CO2 electrolysis at −0.85 V vs. NHE for 24 h. A continuous reaction current of around −0.26 mA was generated (Fig. S3, ESI†). Fig. 2 shows the time-courses of the reduction products and faradaic efficiency (FE) for CO over [Co-dmbpy]. CO was predominantly produced, with trace amounts of H2 and formic acid, whereas other gaseous species such as methane were not detected. The CO generation rate stabilized at a value of ∼5 μmol h−1 after the induction period for 3 h, with the FE for CO formation reaching almost 100% after 5 h of electrolysis (Fig. 2b). As shown in Fig. 2a, the amount of CO (green) produced increased almost linearly, approximately consistent with the ideal value calculated from the number of electrons measured (orange) for the two-electron reduction reaction. The CO selectivity among the total reduction products (CO, H2, and HCOO−) in 24 h reached 99.8%. The turnover number was 3.0, suggesting that this CO2RR was catalytic. When electrolysis was terminated, the formation of gas-phase components quickly ceased. These results show that a stable and highly CO2-selective catalyst originated from [Co-dmbpy] after a certain induction time. No reduction products were detected in electrolysis under the same conditions (−0.85V vs. NHE, pH 6.8) in an Ar atmosphere (Fig. S4, ESI†), suggesting that CO formation is due to CO2 reduction. In the UV-Vis spectrum of the CO2 electrolysis solution, the peaks originating from π–π* transitions of the Co complex showed almost no change between before and after 24 h of electrolysis, suggesting that the Co component was still present as a water-soluble complex in the solution even after the CO2RR (Fig. S5, ESI†). No evidence originating from heterogeneous species of Co components was detected on the CP electrode after 24 h of electrolysis, suggesting that the homogeneous species in the solution were driven as catalysts (Fig. S6 and S7, ESI†). From CO2 electrolysis at different potentials, the overpotential for CO generation was estimated to be 270 mV relative to the theoretical value19,20 (Table S1, entry 1 and Fig. S8, ESI†). Thus, [Co-dmbpy] achieved greater CO selectivity at lower potentials during CO2 electrolysis than previously reported water-soluble molecular catalysts with polypyridine ligands (Table S2, ESI†). The CO selectivity varied significantly with the applied potential. For example, in the CO2 electrolysis at −1.3 V vs. NHE (at pH 6.8), near the second reductive peak in the CV, competitive H2 production was more predominant than CO production (Table S1, entry 4 and Fig. S9, ESI†). This result indicates that the CO2RR proceeds at a more positive potential than that for H2 production over [Co-dmbpy], despite the reaction occurring in an aqueous medium, as shown in the CV curves acquired under Ar and CO2 (Fig. 1b).
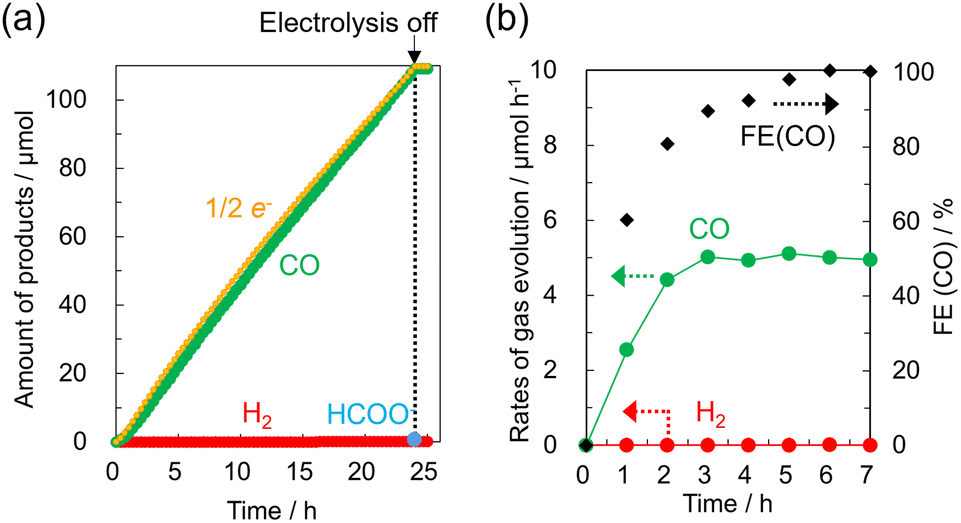 | ||
| Fig. 2 CO2 electrolysis of [Co-dmbpy] (0.3 mmol L−1) in an aqueous NaHCO3 (0.1 mol L−1, 120 mL) solution at −0.85 V vs. NHE (pH 6.8) under CO2 flow (1 atm). (a) Time-course of the total product amounts of CO and H2 (the HCOO− content in the solution was measured at 24 h); 1/2 e− represents the theoretical total amount of two-electron reduction products calculated from the current observed at 100% current efficiency. (b) Time-dependent rates of initial CO2 electrolysis for gas evolution and the faradaic efficiency (FE) for CO. | ||
To measure the change in the electronic state of Co in the aqueous solution at the cathode side, we carried out operando X-ray absorption spectroscopy (XAS) measurements via a fluorescence detection method (Fig. S2, ESI†). XAS measurements of Co complexes in such a dilute (0.5 mmol L−1) aqueous solution have not been reported. As shown in the Co K-edge X-ray absorption near edge structure (XANES) spectra originated from [Co-dmbpy] in Fig. 3a, immediately after a potential was applied (as an integrated signal for 0–20 min), the energy position of half-value of the normalized absorbance of the Co K-edge energy shifted negatively by 1.5 eV and the profile was stabilized even after electrolysis for 3 h. Another operando setup that allowed ∼90% of Co K-edge X-ray signal to be attributed to the CP vicinity also yielded an XANES peak shift similar to that in Fig. 3a (Fig. S10 and S11, ESI†). These significant shifts are due to electron injection into Co complexes at −0.85 V vs. NHE, which might accompany a structural change to an active catalyst. Fig. 3b shows the proposed mechanism for estimating the electrochemical CO2RR by [Co-dmbpy] based on the density functional theory (DFT) calculations. The calculations suggested that a ligand-decoordinated species of [Co(dmbpy)2]2+(1) is formed as an active monomer catalyst by the decoordination of one dmbpy ligand in [Co-dmbpy]. The energy diagram results based on the calculations for the two-coordinated complex (Fig. S12, ESI†) indicate that the highest energy barrier in the scheme of Fig. 3b is the process from 2 → 3 (CO2 coordination), which is the rate-limiting step in the overall CO2RR. Therefore, it is considered that the −1.5 eV shift in the operando XANES spectra is attributable mainly to the active species 2.
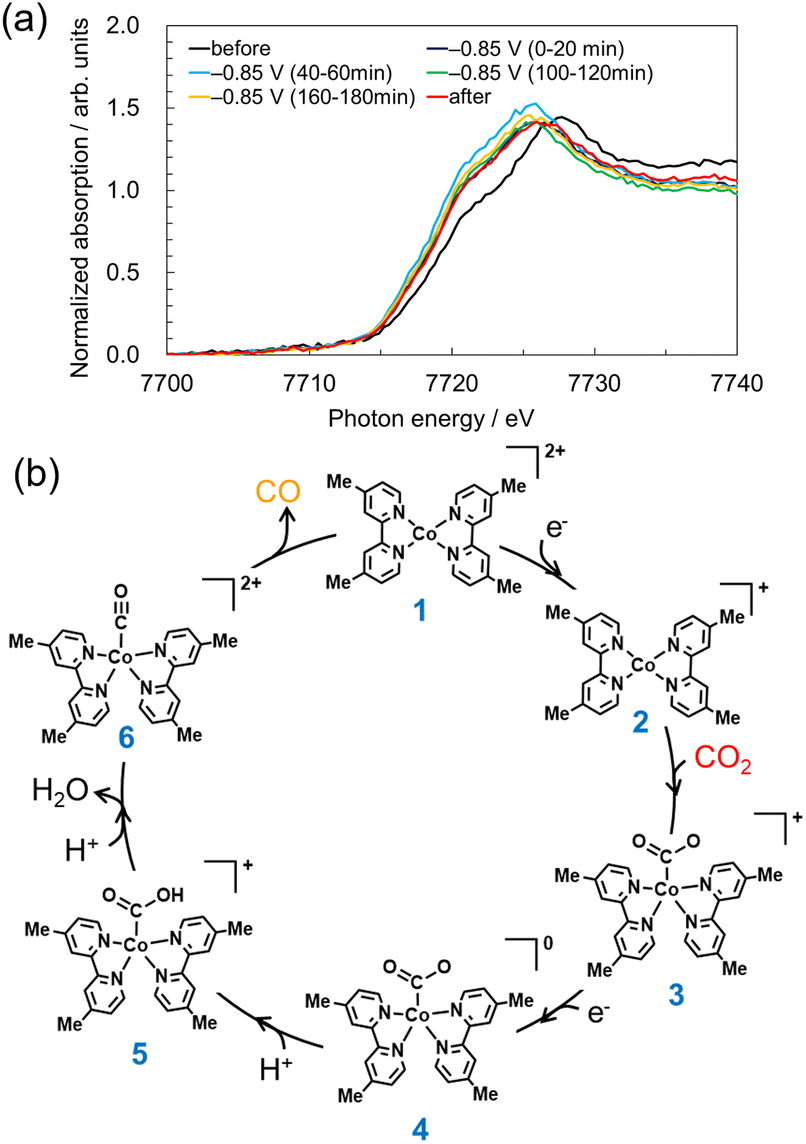 | ||
| Fig. 3 (a) Operando Co K-edge XANES spectra of 0.5 mmol L−1 [Co-dmbpy] measured at open circuit (OC) voltage (before: black), CO2 electrolysis (−0.85 V vs. NHE at pH 6.8), and after the electrolysis measured at OC voltage (after: red) measured in an aqueous 0.1 mol L−1 NaHCO3 solution bubbled with CO2. (b) Proposed electrocatalytic reduction mechanism for CO2 to CO by two-coordinated complex 1 in an aqueous solution based on DFT calculation. | ||
To verify the above hypothesis, we synthesized a pseudo-two-coordinate complex by mixing Co2+ and twice the amount (equivalent) of dmbpy (hereafter, [Co/dmbpy = 1/2]), and we performed CO2 electrolysis using it as a catalyst (Fig. S13–S15, ESI†). The induction period for CO formation by [Co/dmbpy = 1/2] was reduced to less than half that for the three-coordinated [Co-dmbpy] (Fig. S15, ESI†). These results suggest that the active species could be the two-coordinated species and the three-coordinated species is most likely a pre-catalyst.
Table 1 summarizes the results of the electrochemical CO2RR for three types of Co complexes having bpy-based ligands with different electron-donating substituents, [Co-tpy], and Co2+ ions but without any ligands, as measured at −0.85 V vs. NHE for 6 h in NaHCO3 aqueous solution (the corresponding structures are shown in Fig. 1a). First, in the case of an aqueous NaHCO3 solution with no Co components, the generation rates for CO and H2 at 6 h were very low: 0.54 and 0.28 μmol h−1, respectively (entry 8). These low rates were attributed to the reaction at the bare CP electrode as previously reported.11 Only hydrogen was produced by Co2+ ions without ligands preferred for CO2 adduct formation (entry 7), indicating that the Co complexes function as a CO2RR catalyst. By contrast, the [Co-dmbpy] produced CO and H2 rates of 5.13 and 0.01 μmol h−1, respectively, along with a small amount (0.1 μmol) of HCOO−; the selectivity toward CO among the total amount of products reached 99.6%, and the catalyst even suppressed H2 generation at the carbon substrate (entry 1). [Co-bpy] and [Co-dmobpy] (dmobpy: 4,4-dimethoxy-2,2-bipyridine) produced CO at rates of 2.01 and 3.72 μmol h−1 with CO selectivities of 86.8% and 95.8%, respectively (entries 3 and 4). Commercially available complexes [Co(II)-bpy] and [Co(III)-bpy] with Cl− as the counter ion (entries 5 and 6) also exhibited activities similar to that for the synthesized [Co(II)-bpy] with NO3− as the counter ion (entry 3). In particular, [Co-dmbpy] and [Co-dmobpy] with electron-donating ligands on bpy yielded high CO selectivity greater than 95%. This result is consistent with the results of the DFT calculations: the electron-donating groups increase the electron density at the Co center, resulting in stronger bonding with CO2 and promoting the CO2RR.18 In the case of [Co-tpy], the rate of CO generation was 0.73 μmol h−1 and the CO selectivity was 57.2%, which was inferior to the performance of Co complexes with bpy-based ligands (entry 2). This result is consistent with the observation that the Z-schematic CO2RR rate using the [Co-tpy] was low in a previous study.21 [Co-tpy] might require much more negative potential to activate by decoordination of a tpy ligand, as suggested in a previous study that included an experiment conducted in DMF–H2O containing tetrabutylammonium perchlorate (TBAP) electrolyte.13
| Entry | Co compound | Current, 6 h/mA | Rate of products, 6 h/μmol h−1 | CO selectivity/%b | |
|---|---|---|---|---|---|
| CO | H2 | ||||
| a Carbon paper, Ag/AgCl, and Pt wire were used as working, reference, and counter electrodes, respectively. The pH of the 0.3 mmol L−1 Co compound/aqueous NaHCO3 (0.1 mol L−1) solution under CO2 (1 atm) was approximately 6.8. Electrolysis was performed for 6 h, and the production rate at 6 h is shown. b Calculated from the rate of CO generation in relation to the total amount of reduced products (CO, H2, and HCOO−) for 6 h. c Commercial product; [Co(II)(bpy)3]Cl2·5H2O. d Commercial product; [Co(III)(bpy)3]Cl3·4H2O. | |||||
| 1 | [Co-dmbpy] | −0.26 | 5.13 | 0.01 | 99.6 |
| 2 | [Co-tpy] | −0.16 | 0.73 | 0.52 | 57.2 |
| 3 | [Co-bpy] | −0.12 | 2.01 | 0.19 | 86.8 |
| 4 | [Co-dmobpy] | −0.21 | 3.72 | 0.11 | 95.8 |
| 5 | [Co(bpy)3]Cl2c | −0.11 | 1.64 | 0.14 | 87.1 |
| 6 | [Co(bpy)3]Cl3d | −0.11 | 1.43 | 0.16 | 92.6 |
| 7 | Co(NO3)2 | −0.92 | 0.00 | 20.2 | 0.0 |
| 8 | None | −0.055 | 0.54 | 0.28 | 65.9 |
We here discuss the reason why CO2 electrolysis of an aqueous Co complex solution occurred even at a low potential equivalent to −0.80 V vs. NHE. With respect to the mechanism of the lowered CO2RR potential over the Co porphyrin, DFT calculations suggest that water facilitated CO2 reduction through a concerted pathway involving a hydrated metal complex.22 DFT calculations and experiments have revealed that a reaction at the Mn-bpy, in harmony with metal cations and the carbon substrate, changes the CO2RR from an endothermic to an exothermic reaction.8,23 In Re complexes, a mechanism due to dimerization has been proposed for the formation of CO in CO2 reduction at high potential regions based on the one-electron pathway, which may also be the case in this system, but it is not evident.6 Further detailed mechanistic analyses are important, and progress based on the investigations is expected for the present water-soluble [Co-dmbpy] system operating at the low potential regions.
The present [Co-dmbpy] yields CO with nearly 100% selectivity at a low potential of −0.80 V vs. NHE (pH 6.8). In the Z-schematic CO2RR photocatalysis, [Co-dmbpy] was coupled with the (CuGa)0.3Zn1.4S2 semiconductor to produce CO with 98% selectivity under visible-light irradiation.18 The conduction-band minimum (CBM) of (CuGa)0.3Zn1.4S2 is located at a substantially more negative position (−1.66 V vs. NHE at pH 5.9). In a femtosecond transient spectroscopy study of a hybrid photocatalyst composed of an N-doped Ta2O5 (N-Ta2O5) semiconductor linked with a Ru complex having bpy ligands [Ru-bpy], electron injection to the [Ru-bpy] not only from the CBM but also via shallow traps of the N-Ta2O5 semiconductor was confirmed.24 Thus, electron injection from shallow traps of (CuGa)0.3Zn1.4S2 to the two-coordinate Co complex is also expected to produce CO (Fig. S16, ESI†). Because of the low overpotential for the CO2RR over the [Co-dmbpy] complex, sufficient room exists to apply it to various semiconductors to construct an efficient photosystem.20,25 This possibility warrants further study in the near future.
In summary, a water-soluble Co(II) complex with dimethyl-bipyridine ligands functioned as an unparalleled electrocatalyst to drive the CO2RR in an aqueous hydrogen carbonate solution bubbled with CO2. CO2 reduction to CO occurred at a low potential of −0.80 V vs. NHE (pH 6.8) with a CO selectivity approaching 100%. Because the activity emerges in an aqueous solution without any organic solvents, it is beneficial to CO2 reduction setups that use water as an electron donor in electrocatalysis and photocatalysis.
The operando XAFS data were acquired at the BL33XU beamline at the SPring-8 facility, with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2021A7038, 2021B7038, 2022A7038 and 2022B7038). AK thanks KAKENHI, Grants-in-Aid for Scientific Research (A) 23H00248, for financial support. The authors wish to thank Mr Makoto Kondo for experimental assistance and Dr Shunsuke Sato, Dr Masataka Ohashi, Dr Takeo Arai, and Mr Koutaro Wada for useful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, J. Am. Chem. Soc., 1986, 108, 7461–7467 CrossRef CAS PubMed.
- C. Costentin, M. Robert, J. M. Saveant and A. Tatin, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6882–6886 CrossRef CAS.
- A. Call, M. Cibian, K. Yamamoto, T. Nakazono, K. Yamauchi and K. Sakai, ACS Catal., 2019, 9, 4867–4874 CrossRef CAS.
- Q.-Q. Bi, J.-W. Wang, J.-X. Lv, J. Wang, W. Zhang and T.-B. Lu, ACS Catal., 2018, 8, 11815–11821 CrossRef CAS.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS.
- A. Nakada and O. Ishitani, ACS Catal., 2018, 8, 354–363 CrossRef CAS.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2017, 7, 70–88 CrossRef CAS.
- S. Sato, K. Saita, K. Sekizawa, S. Maeda and T. Morikawa, ACS Catal., 2018, 8, 4452–4458 CrossRef CAS.
- M. Wang, K. Torbensen, D. Salvatore, S. Ren, D. Joulié, F. Dumoulin, D. Mendoza, B. Lassalle-Kaiser, U. Işci, C. P. Berlinguette and M. Robert, Nat. Commun., 2019, 10, 3602 CrossRef PubMed.
- J. J. Walsh, G. Neri, C. L. Smith and A. J. Cowan, Organometallics, 2018, 38, 1224–1229 CrossRef.
- J. Wang, L. Gan, Q. Zhang, V. Reddu, Y. Peng, Z. Liu, X. Xia, C. Wang and X. Wang, Adv. Energy Mat., 2019, 9, 1803151 CrossRef.
- S. P. Luo, L. Z. Tang and S. Z. Zhan, Inorg. Chem. Commun., 2017, 86, 276–280 CrossRef CAS.
- N. Elgrishi, M. B. Chambers, V. Artero and M. Fontecave, Phys. Chem. Chem. Phys., 2014, 16, 13635–13644 RSC.
- Y. Yao, Y. Gao, L. Ye, H. Chen and L. Sun, J. Energy Chem., 2018, 27, 502–506 CrossRef.
- J. Lin, Z. Pan and X. Wang, ACS Sustainable Chem. Eng., 2013, 2, 353–358 CrossRef.
- J. J. Walsh, C. Jiang, J. Tang and A. J. Cowan, PCCP, 2016, 18, 24825–24829 RSC.
- Y. Zhang, M. Cao, H. Feng, D. Liu and Q. Li, ACS Catal., 2023, 13, 11376–11388 CrossRef CAS.
- T. M. Suzuki, S. Yoshino, K. Sekizawa, Y. Yamaguchi, A. Kudo and T. Morikawa, Appl. Catal., B, 2022, 316, 121600 CrossRef CAS.
- S. Yoshino, T. Takayama, Y. Yamaguchi, A. Iwase and A. Kudo, Acc. Chem. Res., 2022, 55, 966–977 CrossRef CAS PubMed.
- H. N. Tian, ChemSusChem, 2015, 8, 3746–3759 CrossRef CAS PubMed.
- T. M. Suzuki, S. Yoshino, T. Takayama, A. Iwase, A. Kudo and T. Morikawa, Chem. Commun., 2018, 54, 10199–10202 RSC.
- K. Miyamoto and R. Asahi, J. Phys. Chem. C, 2019, 123, 9944–9948 CrossRef CAS.
- T. Morikawa, S. Sato, K. Sekizawa, T. M. Suzuki and T. Arai, Acc. Chem. Res., 2022, 55, 933–943 CrossRef CAS PubMed.
- K. Yamanaka, S. Sato, M. Iwaki, T. Kajino and T. Morikawa, J. Phys. Chem. C, 2011, 115, 18348–18353 CrossRef CAS.
- H. Kumagai, G. Sahara, K. Maeda, M. Higashi, R. Abe and O. Ishitani, Chem. Sci., 2017, 8, 4242–4249 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc03940d |
| This journal is © The Royal Society of Chemistry 2023 |