
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzymatic reversion of Pt(II) nucleophilicity through charge dumping: the case of Pt(CN)42−†
Sergi
Burguera
,
Antonio
Frontera
 and
Antonio
Bauzá
*
and
Antonio
Bauzá
*
Departament de Química, Universitat de les Illes Balears, Ctra. de Valldemossa Km 7.5, 07122, Palma de Mallorca, Baleares, Spain. E-mail: antonio.bauza@uib.es
First published on 3rd October 2023
Abstract
Combining computations and X-ray structure analysis we have demonstrated that [Pt(CN4)]2− can behave as a Lewis acid inside an enzyme's cavity. The nature of a counterintuitive contact found between a catalytically active GLN residue belonging to a mitochondrial synthase and the Pt(II) center was investigated by combining molecular dynamics and quantum mechanics calculations. Results confirm the electron acceptor role of [Pt(CN4)]2−, serving as an inspiration for the design of biomolecular cages able to tweak the nucleophilic/electrophilic character of an organometallic compound.
Cardiolipin is a dimeric glycerophospholipid commonly located in the mitochondrial membrane of mammalian cells which participates in several organelle phenomena (e.g., cristae formation, oxidative phosphorylation, mitochondrial protein import, mitophagy and apoptosis).1–4 The biosynthetic routes to obtain cardiolipin require cardiolipin–diacylglycerol (CDP–DAG) as a precursor molecule.5–8 In mammals, the biosynthesis of CDP–DAG is carried out by the translocator assembly and maintenance protein 41 (Tam41), using phosphatidic acid and cytosine triphosphate (CTP) as substrates.9
Recently, Jiao and collaborators10 experimentally determined (X-ray diffraction) the structure of an “L”-shaped pocket from Tam 41 where CTP binds, located between a nucleotidyltransferase (NTase) and a winged helix domain. The authors used K2Pt(CN)4 to assist in the structure determination and a comparison between the two protein–ligand complexes (Tam 41–CTP and Tam 41–K2Pt(CN)4, Fig. 1a and b, respectively) revealed common structural features, since the “L”-shaped pocket acted as a “gate”, regulating the CTP/[Pt(CN)4]2− binding to the active site.
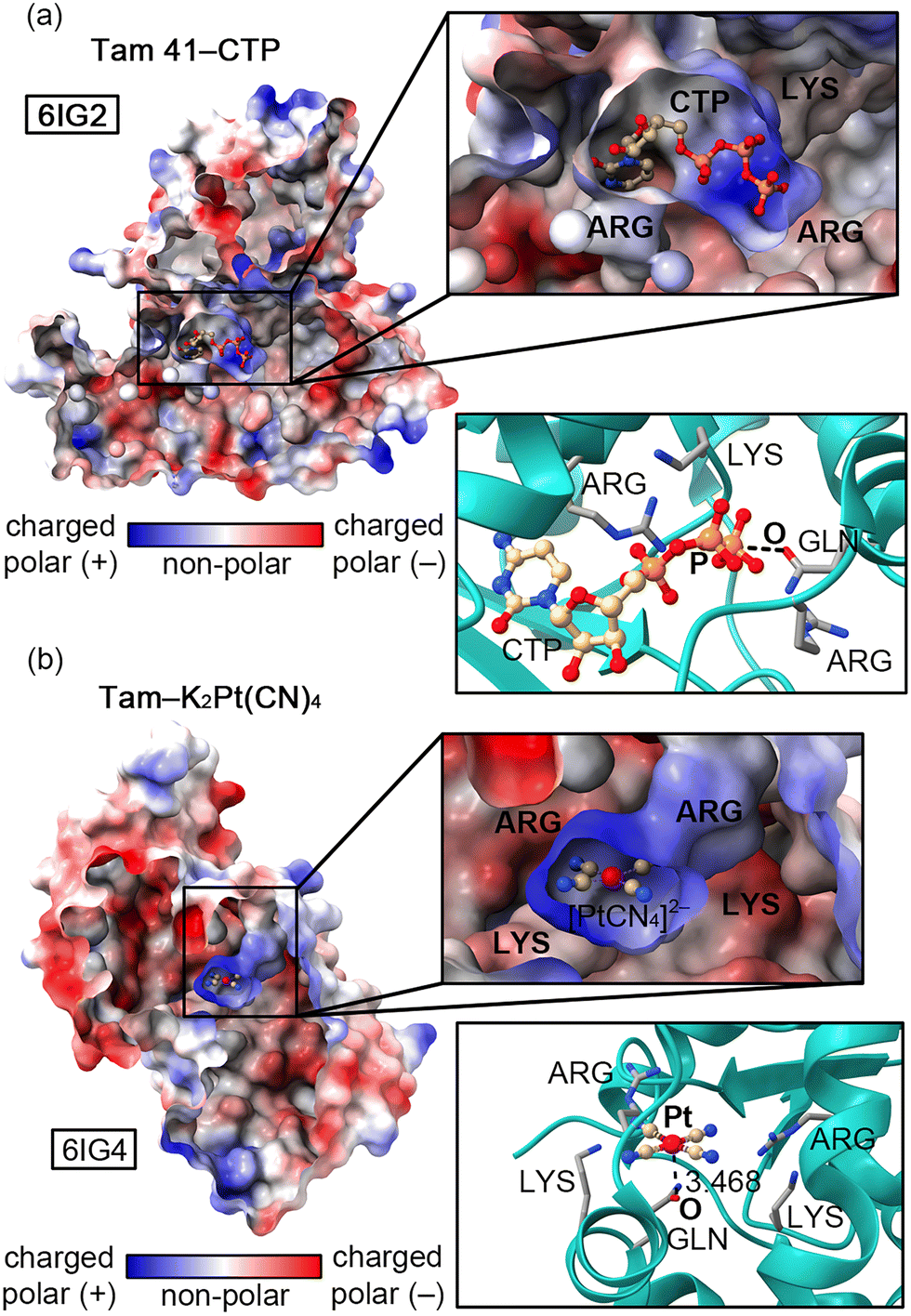 | ||
| Fig. 1 Sliced views of 6IG2 (a) and 6IG4 (b) X-ray structures coloured using the polarity of the amino acids (AAs). The “L”-shaped pocket containing the CTP and [Pt(CN)4]2− species along with the interacting AAs is magnified inside the square parts of the figure. | ||
The authors attributed this behaviour to the presence of two PHE residues located on a vicinal loop (not shown in Fig. 1). The “L” shaped enzymatic cavity is mainly composed by two ARG and two LYS residues that are involved in the recognition of the β- and γ-phosphate groups of CTP through electrostatically enhanced Hydrogen Bonding (HB) interactions.11 Apart from these positively charged residues, a GLN residue is also highly conserved among different Tam41 variants, thus pointing to a common CTP recognition mechanism. In fact, mutation of this residue resulted in a nearly inactive enzyme,12,13 while mutations of LYS and ARG residues reduced the functionality of the wild-type activity to minimal values. Interestingly, this residue is involved in (i) a back bonding Pnictogen Bond (PnB)11,12 interaction with the γ-phosphate group of CTP (Fig. 1a) and (ii) a noncovalent contact with the Pt centre of [Pt(CN)4]2− (Fig. 1b).
While these contacts should be repulsive from an electrostatics perspective (also due to high energy lone pairs located in dz2 and dx2−y2 orbitals that increase the nucleophilicity of the Pt centre) we found a remarkable charge-dumping effect that stabilizes both negatively charged species inside the “L”-shaped pocket, resulting in the appearance of electrophilic regions over the P and Pt atoms, able to interact with the O atom from the GLN residue. As noted in Fig. 2, a significant variation in the electrostatic potential over the P/Pt atom was found upon the formation of HBs with the four cationic species (formamidinum and ammonium cations were used as a theoretical approximation to mimic the ARG and LYS residues present in the enzyme's pocket), changing from −318.7 to −38.3 kcal mol−1 in the case of CTP (Fig. 2a) and from −168.2 to +72.8 kcal mol−1 in the case of [Pt(CN)4]2− (Fig. 2b). A similar situation has been described in both experimental14 and computational studies,15,16 where a metal atom belonging to an anionic moiety (perrhenate anion or tetracyanidonitridotechnetium dianion) surrounded by cationic species acted as electron acceptor (Matere Bond). In addition, square planar tetracoordinated aluminum(III) anions have also shown an electrophilic behaviour when put in a proper chemical environment.17 Lastly, our group has reported a similar dumping effect for NO3−.18 Despite these previous works, this effect has not been described for an organometallic compound present in an enzyme's pocket.
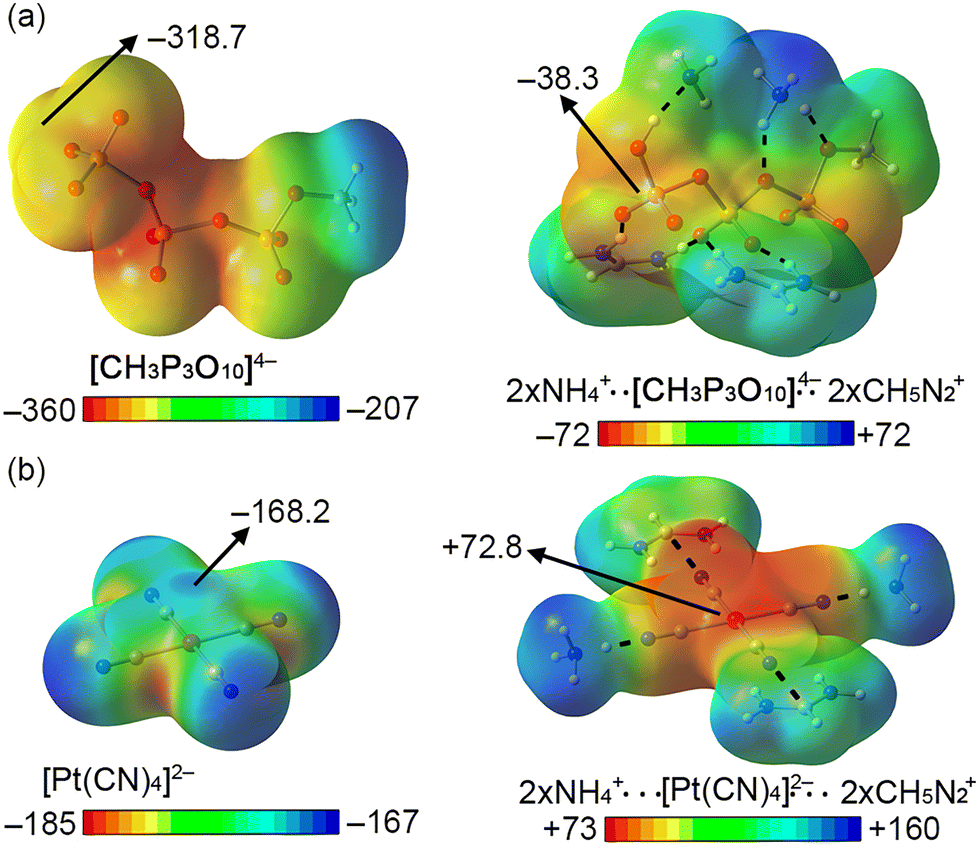 | ||
| Fig. 2 Molecular Electrostatic Potential (MEP) analysis of the [CH3P3O10]4−/[Pt(CN)4]2− (a) and [CH3P3O10]4−/[Pt(CN)4]2− interacting with two NH4+ and two CH5N2+ molecules (b). HBs indicated using black dashed lines. Energy values at selected points in the surface are given in kcal mol−1 (0.002 a.u.). | ||
To investigate the stabilizing role of the four positively charged residues that compose the “L”-shaped pocket, MD (Molecular Dynamics) simulations using the NVT ensemble (100 ns production in addition to three replicas of 50 ns, see ESI† for specific details regarding the MD protocol and the results from the replicas) of one of the subunits from the Tam 41–[Pt(CN)4]2− complex were carried out. In Fig. 3a, the RMSD (Root Mean Square Deviation) plot of the Pt⋯ARG and Pt⋯LYS distances along the 100 ns trajectory can be observed. Interestingly, ARG148 (coloured in orange) and LYS151 (coloured in blue) residues are the ones that fluctuate the most along the trajectory, showing RMSD values comprised between 8 and 12 Å. Particularly, the RMSD is more stable in the latter while the former shows a larger fluctuation, although between reasonable values.
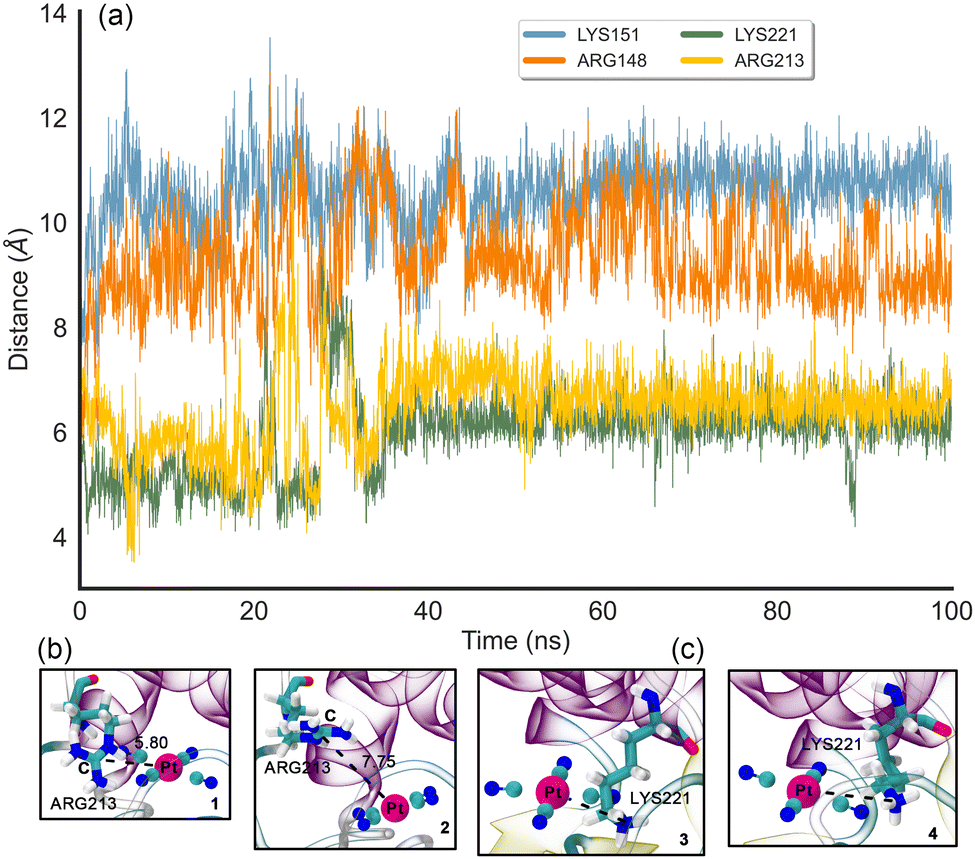 | ||
| Fig. 3 RMSD plot of the (CN)4–Pt−⋯NH3+ and (CN)4–Pt−⋯CH4N2+ distances along the 100 ns MD trajectory using the first frame as reference (a). Distances Pt⋯ARG are measured to the central C atom from the formamidinium group. Distances Pt⋯LYS are measured to the N atom of the ammonium group. Pt⋯ARG213 (regions 1 and 2) and Pt⋯LYS221 (regions 3 and 4) interactions at selected snapshots of the MD trajectory (b) and (c). The square areas in Fig. 3a indicate where the snapshots were taken. | ||
Another worth noting aspect is the significant increase observed in the RMSD for ARG213 (coloured in yellow) and LYS221 (coloured in green) (reaching values near 9–10 Å) between 20 and 40 ns of simulation, which corresponds a back-and-forth tilting motion of the PtCN42− molecule inside the “L”-shaped pocket in the case of ARG213 residue (Fig. 3b). On the other hand, in the case of LYS221, we observed a twisting move of the amino acid sidechain, resulting in an increase of the Pt⋯N distance (Fig. 3c). Once passed the 40 ns of simulation, both Pt⋯ARG/LYS distances fluctuate between closer values (6–8 Å) during the rest of the trajectory, thus tightly stabilizing the anionic moiety through the electrostatically enhanced HBs.
From these analyses, we propose that ARG213 and LYS221 are the residues that contribute the most to the stabilization of the anionic moiety (through a HB-based charge dumping effect) inside the enzymatic cavity, while ARG148 and LYS151 stay farther from the [Pt(CN)4]2− molecule during the simulations, being their HBs weaker and consequently lowering their stabilizing contribution. It is also worth mentioning that the average Pt⋯LYS/ARG values observed in the trajectories and the ones derived from the initial X-ray structure lied within the same range (between 5 and 8.5 Å in the case of the X-ray), with one LYS and one ARG residue much closer to the Pt centre (dPt⋯LYS = 5.282 Å and dPt⋯Arg = 6.338 Å), in line with the results obtained from the simulations. Another interesting aspect is related to the stability of the Pt(II) centre, which remained in a square planar fashion along the entire trajectories, with slight variations in the C–Pt–C angles and C–Pt–C–C dihedrals (see Fig. S4–S6 in the ESI†), thus indicating that the parameters used for modelling the geometry of [Pt(CN4)]2− were correctly implemented.
To shed light into the physical nature of the counterintuitive O⋯Pt contact, Quantum Mechanics (QM) calculations were performed. The understanding of this interaction is of great importance, since this residue is involved in the native substrate recognition as well in the catalytic process of CDP–DAG biosynthesis. In order to achieve this, we conducted an ab initio theoretical study (RI-MP219/def2-TZVP20 level of theory) using several electron rich moieties (C, O, N and S atoms) and the (NH4)2+⋯[Pt(CN)4]2−⋯(CH5N2)2+ system (see Fig. 4). Initially, to demonstrate the electrophilicity of the Pt(II) atom from the [Pt(CN)4]2− complex, a very convenient methodology was used (see Fig. 5 for a concrete example) based on the comparison of the electron density (ED) and the electrostatic potential (ESP) distribution along the Pt(II)⋯A (A![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, N, O and S) bond path.21 In any donor–acceptor interaction, the ED minimum is closer to the electron acceptor atom and the ESP minimum is closer to the electron donor atom.22 The plot shown in Fig. 5 involves complex 2 and the positions of ED and ESP minima along the Pt(II)⋯C–C bond path confirm the electron donation of the π-system involving the C–C bond to the Pt(II), thus revealing that the Pt(II) is acting as the electrophile. The distance between both minima is also indicated in Fig. 5, which was 0.16 Å.
C, N, O and S) bond path.21 In any donor–acceptor interaction, the ED minimum is closer to the electron acceptor atom and the ESP minimum is closer to the electron donor atom.22 The plot shown in Fig. 5 involves complex 2 and the positions of ED and ESP minima along the Pt(II)⋯C–C bond path confirm the electron donation of the π-system involving the C–C bond to the Pt(II), thus revealing that the Pt(II) is acting as the electrophile. The distance between both minima is also indicated in Fig. 5, which was 0.16 Å.
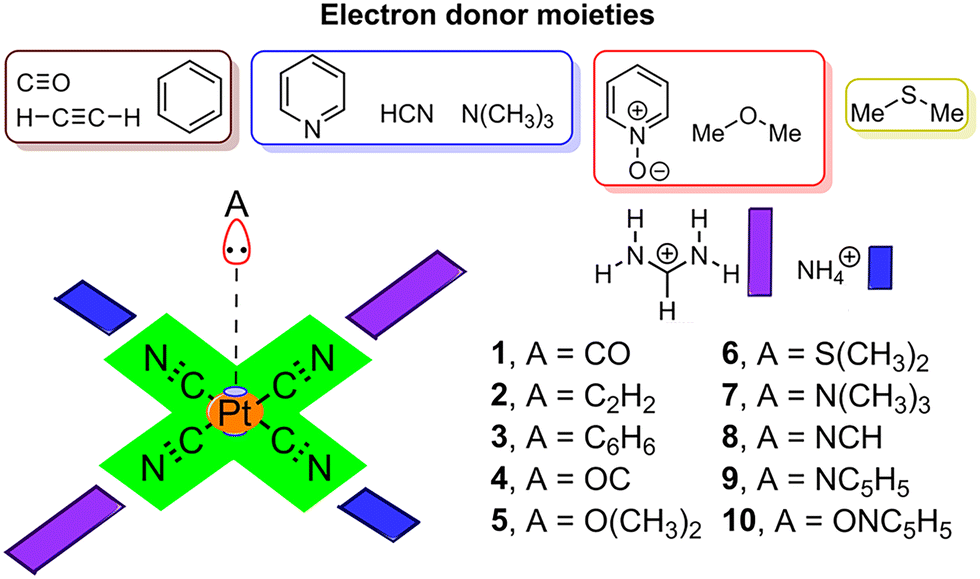 | ||
| Fig. 4 Compounds and complexes 1 to 10 used in this study. | ||
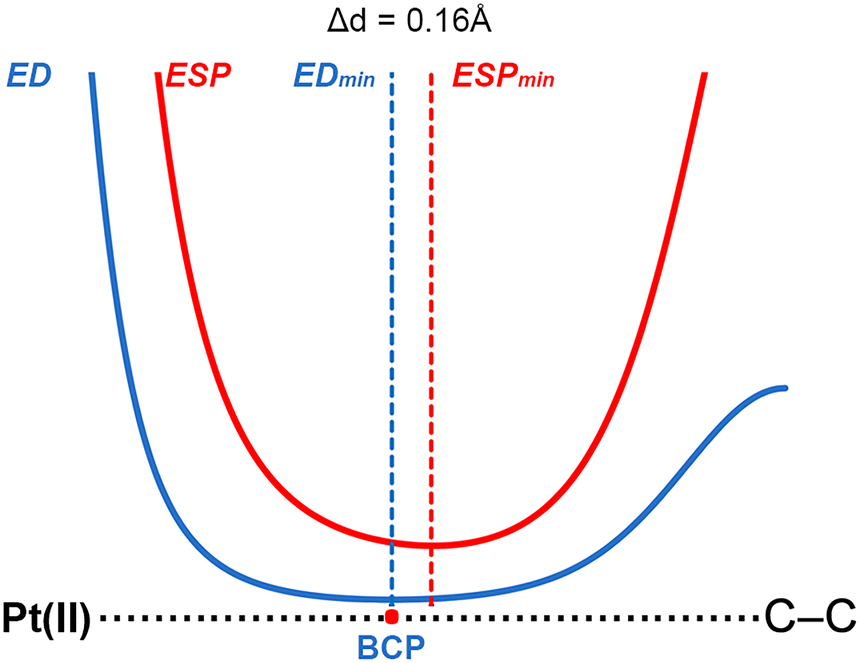 | ||
| Fig. 5 Complex 2 involving ethyne ED/ESP profile along the Pt(II)⋯C–C bond path. | ||
Regarding the energetics of the noncovalent complexes studied (see Table 1), in all the cases favourable interaction energy values were obtained, comprised between −4.3 and −0.5 kcal mol−1. The energies followed the expected trend which is related to the weaker/stronger basicity of the electron donor atom, being those noncovalent complexes involving trimethylamine and pyridine (7 and 9) the most favourable ones (−5.0 and −4.3 kcal mol−1, respectively), followed by complexes 5 and 10 involving dimethylether and pyridine-N-oxide (−2.3 and −3.6 kcal mol−1, respectively). On the other hand, among the C donor molecules complex 3 involving benzene achieved the largest interaction energy value (−4.1 kcal mol−1). In contrast, complex 4 involving OC as electron donor molecule achieved the poorest interaction energy value of the set (−0.1 kcal mol−1). These results agreed with calculations using a theoretical model of 6IG4 structure (see ESI† for the cartesian coordinates), revealing a O⋯Pt interaction strength of −2.2 kcal mol−1.
| Complex | ΔE | ΔEBSSE | d | ρ × 100 |
|---|---|---|---|---|
| 1 (Pt⋯CO) | −1.2 | −0.5 | 3.750 | 0.51 |
| 2 (Pt⋯C2H2) | −2.9 | −1.5 | 3.392 | 1.02 |
| 3 (Pt⋯C6H6) | −7.4 | −4.1 | 3.389 | 0.69 |
| 4 (Pt⋯OC) | −0.6 | −0.1 | 3.619 | 0.42 |
| 5 (Pt⋯O(CH3)2) | −4.9 | −2.4 | 3.038 | 1.47 |
| 6 (Pt⋯S(CH3)2) | −7.5 | −4.4 | 3.082 | 2.71 |
| 7 (Pt⋯N(CH3)3) | −8.9 | −5.0 | 2.798 | 3.04 |
| 8 (Pt⋯NCH) | −3.3 | −2.6 | 3.545 | 0.62 |
| 9 (Pt⋯NC5H5) | −7.0 | −4.3 | 2.826 | 2.71 |
| 10 (Pt⋯ONC5H5) | −5.4 | −3.6 | 3.048 | 1.18 |
We have also analysed this noncovalent contact from the perspective of the charge density theory by computing the QTAIM23 (Quantum Theory of Atoms in Molecules) and NBO24 (Natural Bonding Orbital) analyses (two examples are highlighted in Fig. 6).
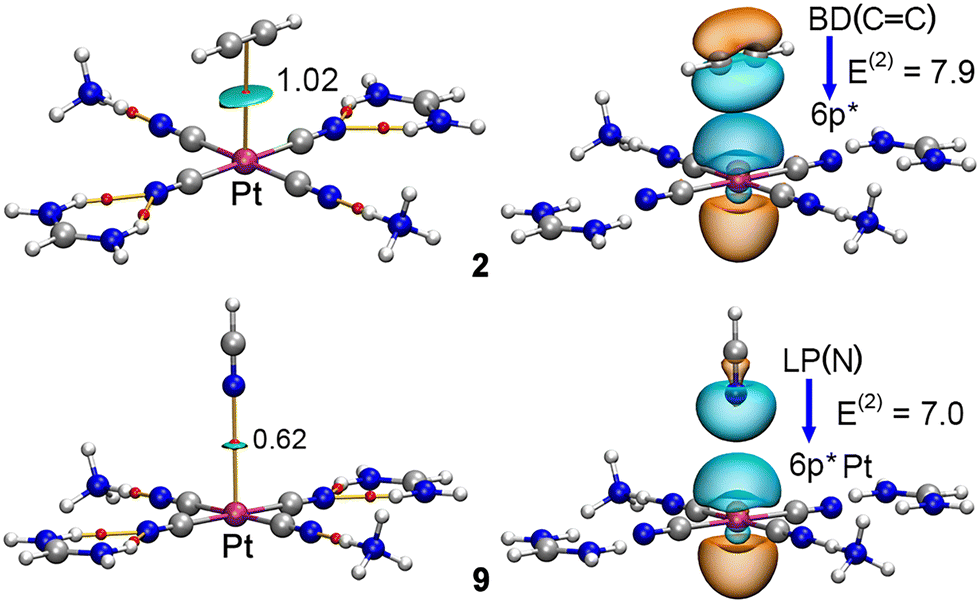 | ||
| Fig. 6 Combined QTAIM and NCIplot analyses of complexes 2 and 9. The NBO graph involving the donor and acceptor orbitals along with the orbital contribution (E(2), in kcal mol−1) is also indicated. | ||
These point out to the presence of a bond critical point (BCP) connecting i) the C–C BCP in complex 2 and (ii) a lone pair of the N atom in complex 9 to the Pt(II) ion. In addition, the weak nature of the interaction was confirmed by the NCIplot25 (Non Covalent Interactions plot) analyses, which exhibited a greenish isosurface located between either the C–C bond or the N atom and the Pt(II) ion. Furthermore, we have also included a graphical representation of the donor and acceptor NBOs that participate in the formation of the Pt⋯C–C/N contacts present in these complexes, confirming the involvement of either a lone pair (LP) or the bonding orbital of the CC π-system [BD(CC)] as donor and an unfilled 6p orbital (6p*) from the Pt(II) ion as acceptor orbitals, respectively, with magnitudes of 7.9 and 7.0 kcal mol−1, respectively. Lastly, we performed the NBO analysis on the 6IG4 model, revealing an orbital contribution from a LP of the O atom to an unfilled 6p orbital (6p*) of the Pt(II) ion with a magnitude of 4.81 kcal mol−1, in line with the results derived from fully optimized complexes. Besides, orbital contributions involving the LP of the sp N atoms from the Pt moiety and antibonding (BD*) H–N orbitals belonging to the cationic moieties were found, with a magnitude of 15.84 kcal mol−1, thus confirming the charge dumping process from an orbital perspective.
In conclusion, we have theoretically described the ability of [Pt(CN)4]2− to behave as a Lewis acid in an enzyme's pocket and counterintuitively interact with an O atom belonging to a catalytically active GLN residue. This was mainly attributed to a noticeably charge-dumping effect due to four vicinal positively charged protein residues (ARG and LYS) that resembles previous reports on inorganic systems. In this regard, MD simulations of one of the protein subunits containing the Pt(CN)42− molecule were useful to account for the stabilizing role of the ARG/LYS residues while ab initio QM calculations at the RI-MP2/def2-TZVP level of theory shed light into the physical nature of the interaction by assigning the donor–acceptor role as well as by unveiling the orbital contributions responsible of the formation of this noncovalent complex. We believe the results reported herein will be useful for the design of bioinspired molecular cavities able to tweak the nucleophilic/electrophilic character of an organometallic compound, which might have significant implications in supramolecular chemistry and catalysis.
This research was funded by the “Ministerio de Ciencia, Investigacion y Universidades/Agencia Estatal de Investigación” (MICIU/AEI) of Spain (project PID2020-115637GB-I00. FEDER funds). We thank the “Centre de les Tecnologies i la Informació” (CTI) and the University of Porto (requimte lab) for computational facilities.
Conflicts of interest
There are no conflicts to declare.References
- R. N. Lewis and R. N. McElhaney, Biochim. Biophys. Acta, 2009, 1788, 2069–2079 CrossRef CAS PubMed.
- S. E. Horvath and G. Daum, Prog. Lipid Res., 2013, 52, 590–614 CrossRef CAS PubMed.
- C. U. Martensoon, K. N. Doan and T. Becker, Biochim. Biophys. Acta, 2017, 1862, 102–113 CrossRef PubMed.
- J. Dudek, Front. Cell Dev. Biol., 2017, 5, 90 CrossRef PubMed.
- S. C. Chang, P. N. Heacock, C. J. Clancey and W. Dowhan, J. Biol. Chem., 1998, 273, 9829–9836 CrossRef CAS PubMed.
- C. Osman, M. Haag, F. T. Wieland, B. Brugger and T. Langer, EMBO J., 2010, 29, 1976–1987 CrossRef CAS PubMed.
- S. C. Chang, P. N. Heacock, E. Mileykovskaya, D. R. Voelker and W. Dowhan, J. Biol. Chem., 1998, 273, 14933–14941 CrossRef CAS PubMed.
- G. Tuller, C. Hrastnik, G. Achleitner, U. Schiefthaler, F. Klein and G. Daum, FEBS Lett., 1998, 421, 15–18 CrossRef CAS PubMed.
- Y. Tamura, Y. Harada, S. Nishikawa, K. Yamano, M. Kamiya, T. Shiota, T. Kuroda, O. Kuge, H. Sesaki, K. Imai, K. Tomii and T. Endo, Cell Metab., 2013, 17, 709–718 CrossRef CAS PubMed.
- H. Jiao, Y. Yin and Z. Liu, Structure, 2019, 27, 1258–1269 CrossRef CAS PubMed.
- S. C. C. van der Lubbe and C. Fonseca Guerra, Chem. – Asian J., 2019, 14, 2760–2769 CrossRef CAS PubMed.
- A. Varadwaj, P. R. Varadwaj, H. M. Marques and K. Yamashita, Inorganics, 2022, 10, 149 CrossRef CAS.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Chem. Phys. Chem., 2016, 17, 1608 CrossRef PubMed.
- Y. Xu, M. Calabrese, N. Demitri, A. Pizzi, T. Nag, I. Hung, Z. Gan, G. Resnati and D. L. Bryce, Chem. Commun., 2023 10.1039/D3CC04090A.
- S. Burguera, R. M. Gomila, A. Bauzá and A. Frontera, Crystals, 2023, 13, 187 CrossRef CAS.
- A. Daolio, A. Pizzi, G. Terraneo, M. Ursini, A. Frontera and G. Resnati, Angew. Chem., Int. Ed., 2021, 133, 14506–14510 CrossRef.
- F. Ebner, H. Wadepohl and L. Greb, J. Am. Chem. Soc., 2019, 141, 18009–18012 CrossRef CAS PubMed.
- A. Bauzá, A. Frontera and T. J. Mooibroek, Nat. Commun., 2017, 8, 14522 CrossRef PubMed.
- F. Weigend and M. Häser, Theor. Chem. Acc., 1997, 97, 331 Search PubMed.
- A. Schaefer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef CAS.
- I. Mata, E. Molins, I. Alkorta and E. Espinosa, J. Phys. Chem. A, 2007, 111, 6425 CrossRef CAS PubMed.
- E. V. Bartashevich, Y. V. Matveychuk, E. A. Troitskaya and V. G. Tsirelson, Comput. Theor. Chem., 2014, 1037, 53 CrossRef CAS.
- R. F. W. Bader, Acc. Chem. Res., 1985, 1, 9 CrossRef.
- F. Weinhold, J. Comput. Chem., 2012, 15, 2363 CrossRef PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Theoretical methods, additional analysis related to molecular dynamics simulations and Cartesian coordinates of complexes 1 to 10 and of the PDB models used. See DOI: https://doi.org/10.1039/d3cc03816e |
| This journal is © The Royal Society of Chemistry 2023 |