
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethane dehydroaromatization catalyzed by Mo/ZSM-5: location-steered activity and mechanism†
Guanna
Li
 ab
ab
aBiobased Chemistry and Technology, Wageningen University, Bornse Weilanden 9, 6708 WG, Wageningen, The Netherlands. E-mail: guanna.li@wur.nl
bInorganic Systems Engineering, Department of Chemical Engineering, Delft University of Technology, Van der Maasweg 9, 2629 HZ, Delft, The Netherlands
First published on 15th August 2023
Abstract
This work examined the location-steered catalytic behavior of Mo/ZSM-5 catalyst for one-step methane dehydroaromatization to benzene reaction. The results indicated that α-site is the preferred location for the formation of ethylene, the main intermediate for aromatics production via the propagation pathway, while δ-site is favorable for the hydrocarbon pool aggregation reaction pathway.
Nonoxidative direct methane dehydroaromatization (MDA) to benzene and hydrogen was first reported in the 1980s.1 Since then many different catalysts have been studied, and the results have indicated that Mo/ZSM-5 is the most promising heterogeneous catalyst compared to other materials.2 However, Mo/ZSM-5 hasn’t drawn much attention from the academic research community in the past few decades because of the fast catalyst deactivation and the lack of advanced characterization techniques to get insight into the nature of the active site and the coke formation process under the harsh reaction conditions.3 In recent years, this catalytic system has come back into the spotlight again because of the development of advanced in situ and operando techniques.4
Regarding the nature of the Mo active site, it has been identified to be Mo(oxy)carbide species formed at the initial stage of reaction by reducing the fresh Mo(VI)-oxo precursors with the reactant of methane.5 The real catalytic MDA reaction is triggered over the so-formed Mo(oxy)carbide sites. This process is accompanied by gradual coke deposition and catalyst deactivation.6 The fundamental issues of the structure of the Mo(oxy)carbide species and the reaction mechanism of the initial methane C–H bond activation and aromatics formation have been investigated by both experimental characterizations and computational modelling.7,8 The general conclusion is that highly dispersed small Mo(oxy)carbide species, particularly mononuclear and binuclear MoCxOy species, confined in the channels of ZSM-5 are responsible for the catalytic MDA reaction.
For the reaction mechanism, two pathways are under intensive investigation by comprehensive in situ and operando characterizations.9 In the bifunctional mechanism, it was claimed that the key intermediate of ethylene is first formed on the Mo active site by C–H bond activation and C–C coupling, which is further propagated to aromatic products over the Brønsted acids.10 Alternatively, Kosinov et al. proposed a hydrocarbon pool reaction pathway, i.e., methane firstly aggregates into polyaromatic carbon species, from which benzene is derived from the secondary reaction between it and the initial product of methane activation.11 The principle underlying this pathway resembles the mechanism established in the methanol-to-olefin reaction. This mechanism was also proved by computational modelling to be kinetically more favourable than the bifunctional mechanism via the ethylene pathway over the δ-site of Mo/ZSM-5.8
Tailoring the micro-environment around the catalytically active sites has a significant influence on the reactivity of various catalysts.12 It was recently reported that zeolitic matrix constraints and the location of the active site can activate and tune the function of metal-containing species.13 Inspired by this research, in this study, computational modelling was performed to assess the effect of distinct local environments of the ZSM-5 framework on the reactivity of the binuclear Mo-carbide species. Various locations in the main channel and zigzag channel of ZSM-5 were selected to stabilize the active site, and both ethylene and hydrocarbon pool reaction pathways were examined. The aim of this work is to explore the influence of different local confinements on the reactivity and mechanism and to identify the most favourable sitting position of the Mo-carbide active site.
Fig. 1 shows the topology of the ZSM-5 framework. Four different locations of α-, β-, γ-, and δ-sites were selected for accommodating the active site, with α- and γ-sites being inside the main channel and zigzag channel, and β- and δ-sites at the intersection between the main and zigzag channels, respectively. The active site is a binuclear Mo2C2 molybdenum carbide cluster charge compensated by two Al sites at each location. This cluster was predicted to be the relevant species for MDA reaction by our previous work. For each location, the aforementioned two reaction pathways, i.e., ethylene and hydrocarbon pool routes, were examined.
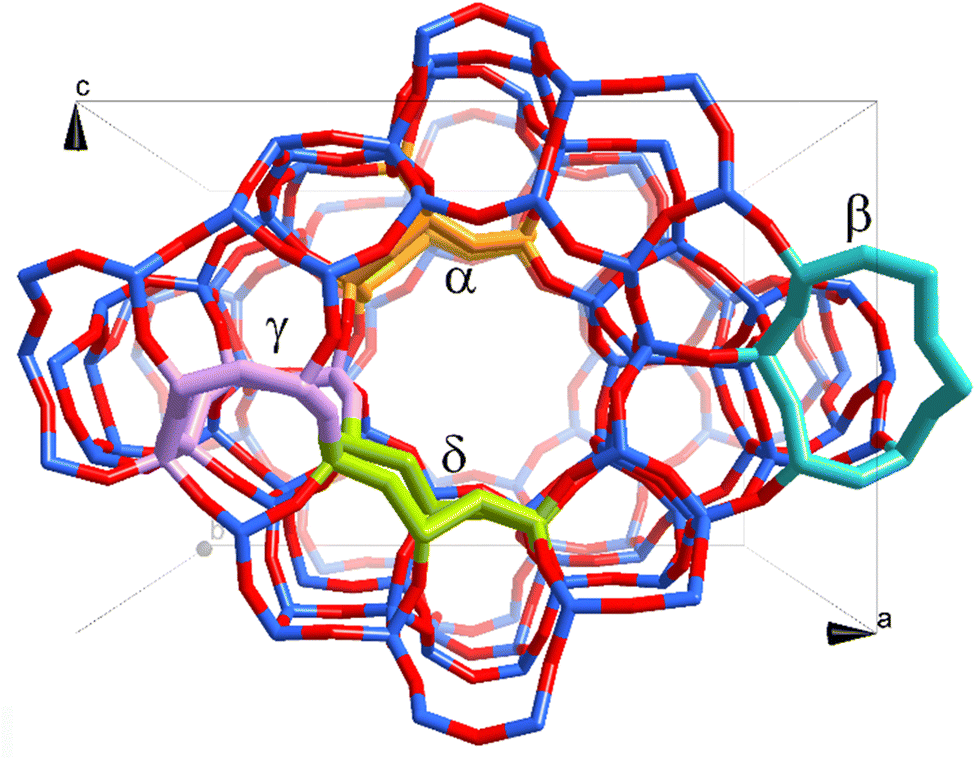 | ||
| Fig. 1 ZSM-5 framework and selected locations to stabilize the Mo carbide site. | ||
The reaction energy profiles over the α-site are demonstrated in Fig. 2. Local geometries are shown in Fig. S8 in the ESI.† The adsorption energy of methane to the active site is −0.57 eV, including the van der Waals interaction with the zeolite framework. Regarding the ethylene formation reaction pathway, the activation of the first C–H bond of methane proceeds heterolytically with an activation barrier of 0.64 eV. The CH3 group is bonded to the Mo site, and hydrogen goes to one of the C sites. This step is exothermic with a reaction energy of 0.48 eV. Further dehydrogenation of the methyl group to form methylene needs to overcome an activation barrier of 1.46 eV. The migration of the so-produced H species from C sites to the Mo site to form a H2 molecule has a barrier of 1.23 eV and 0.97 eV, respectively. Desorption of the H2 molecule to the gas phase is endothermic, but it should be thermodynamically favorable considering the significant entropic compensation at the high reaction temperature. The adsorption of the second methane molecule is also favorable with an adsorption energy of −1.0 eV. Subsequently, a similar reaction of methane C–H bond dissociation takes place, with H atom binding to the Mo site and the methyl group to the C site. Then the C–C coupling reaction can occur between the methylene and the methyl groups to form a C2H5 ethyl group. The activation barrier of this reaction step is 1.86 eV. Ethylene is formed by one further step of dehydrogenation reaction of the C2H5 moiety, with the H atom going to the C site. The barrier of this step is only 0.5 eV. Finally, the H species migrates to the Mo site to form another H2 molecule. The catalytic cycle is closed by the co-desorption of ethylene and H2 molecules from the Mo2C2 site into the gas phase. The desorption energy of this step is 2.99 eV.
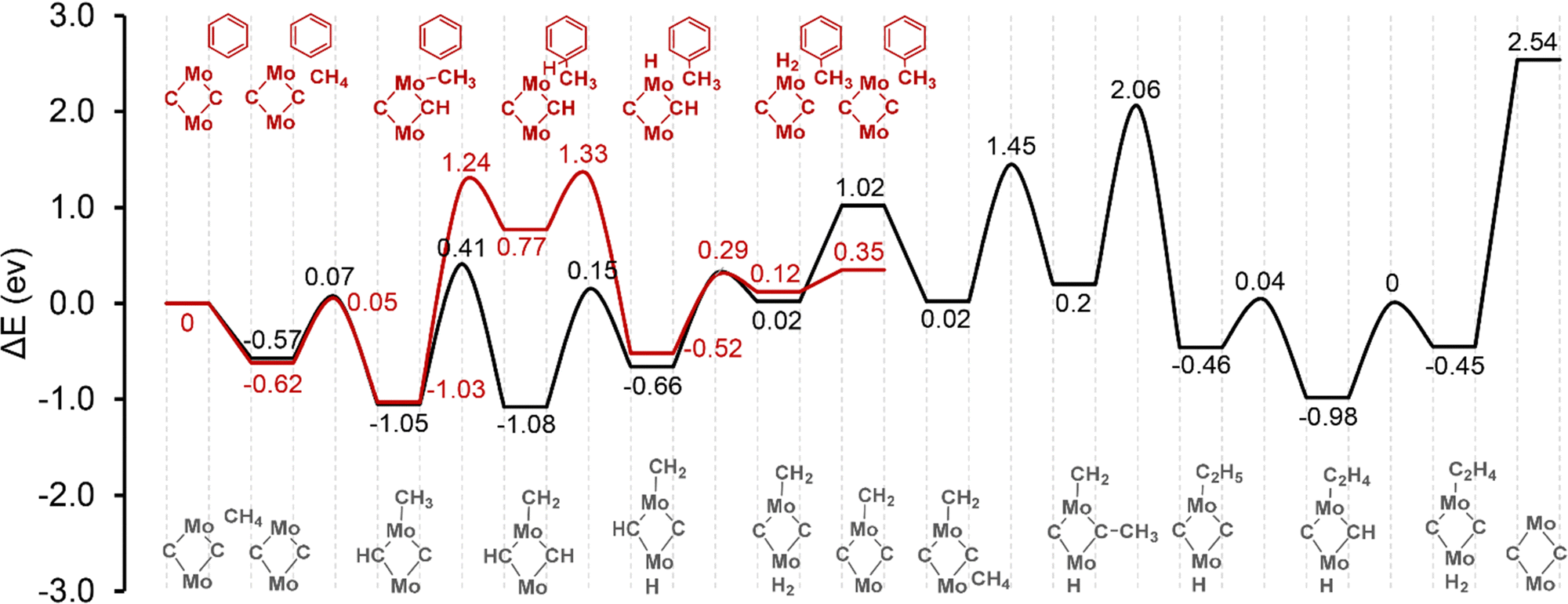 | ||
| Fig. 2 Reaction energy diagram of methane activation over [Mo2C2]2+ located at the α-site. Color codes: red – ethylene formation pathway; black – hydrocarbon pool pathway. | ||
To investigate the hydrocarbon pool mechanism, the benzene molecule present next to the Mo2C2 active site is adopted as a model hydrocarbon pool polyaromatic species. It is shown that both the ethylene and hydrocarbon pool reaction pathways share very similar thermodynamics and kinetics for methane adsorption and the first C–H bond activation reactions. The methylation of benzene to form a C7H9˙ complex is endothermic with an activation barrier of 2.27 eV and a reaction energy of 1.8 eV. The transfer of an H atom from the C7H9˙ complex back to the neighboring Mo site is thermodynamically favorable. It shows an activation barrier of 0.56 eV and a reaction energy of −1.29 eV. The recombination of the two H species at Mo and C sites to form a H2 molecule needs to overcome a barrier of 0.81 eV and the desorption of the H2 molecule from the active site of Mo2C2 only has a barrier of 0.23 eV. From these results, it can be seen that C–C bond formation over the Mo2C2 site is the most difficult step for methane to ethylene reaction, while the benzene methylation step has the highest activation barrier for the hydrocarbon pool route.
The influence of Mo2C2 locations on the activity and MDA reaction mechanism was evaluated by comparing methane activation over the α-site with other sites of β, γ and δ. For each location, two distinct pathways via ethylene and hydrocarbon pool were evaluated (Fig. S1–S3, ESI†). Over the β-site, the methane adsorption energy is −0.36 eV and the activation barrier for the first C–H bond dissociation is 0.75 eV. Dehydrogenation of methyl to methylene has an activation barrier of 1.42 eV. The barriers for the migration of a H atom from the C site to the Mo site and the recombination of two H atoms to form a H2 molecule are 1.79 eV and 1.06 eV, respectively. The desorption energy of a H2 molecule from the Mo2C2 site to the gas phase is 0.7 eV. Adsorption of the second methane molecule to the active site stabilizes the system by 0.31 eV. The activation barrier for the first C–H bond dissociation is 0.93 eV. The C–C bond formation is again the most difficult reaction step with an activation barrier of 2.99 eV. The migration and recombination of the two H species over the active site to form a H2 molecule is a very easy process with barriers of 0.15 eV and 0.75 eV, respectively. The desorption of ethylene and H2 to the gas phase has a barrier of 2.53 eV.
Methane adsorption over the β-site becomes endothermic (0.08 eV) when the benzene molecule is present adjacent to the Mo2C2 site. The C–H bond dissociation needs to overcome a barrier of 0.63 eV. The next step of benzene methylation to form the C7H9˙ compound needs to overcome a barrier of 2.35 eV. The reaction energy for this step is 1.3 eV. Dehydrogenation of the C7H9 compound has a barrier of 0.61 eV and a reaction energy of 1.37 eV. Formation of the H2 molecule is relatively straightforward. The barrier for H atom shifting from the C to Mo site is 0.93 eV, while the desorption of the so-formed H2 is thermodynamically favorable.
Fig. S2 (ESI†) demonstrates the reactivity of Mo2C2 species located at the γ-site. The adsorption energy of a methane molecule is −0.52 eV. The activation barrier of the first C–H bond cleavage is 0.81 eV. Further dehydrogenation of the methyl group to methylene has an activation barrier of 0.88 eV, and a reaction energy of −1.48 eV. Methylene formed over the γ-site is much more stable than that over the α- and β-sites. Formation of the first H2 molecule is energetically not favorable. The desorption barrier of H2 is 0.87 eV. The adsorption energy of the second methane over the active site is 0.97 eV. The C–H bond dissociation has a barrier of 1.24 eV. The most difficult reaction step of C–C bond formation needs to overcome a barrier of 2.42 eV. Once the ethyl group is formed, the subsequent dehydrogenation and H2 formation steps can take place smoothly with minor barriers. The last step of ethylene and H2 desorption and Mo2C2 active site regeneration has a barrier of 2.99 eV at the location of the γ-site.
Similar to what is observed over the other sites, the activity of Mo2C2 for methane C–H bond cleavage is very similar for both the ethylene and hydrocarbon pool pathways. The methylation reaction needs to overcome an activation barrier of 2.85 eV, with a reaction energy of 1.82 eV, which is the most difficult step. Once this barrier is overcome, the following steps of the dehydrogenation of the C7H9˙ compound and the formation of H2 are much easier and can proceed with moderate obstacles. The activation barrier of C7H9˙ dehydrogenation is only 0.71 eV and the barrier for the two H atom recombinations is 1.14 eV.
The fourth sitting position studied is the δ-site (Fig. S3, ESI†), which was predicted to be the most stable location for the stabilization of the Mo2C2 species.8 The adsorption energy of methane over the active site is −0.57 eV. The activation barrier of the first C–H bond dissociation is 0.57 eV and the reaction energy is −0.62 eV. Dehydrogenation of the methyl group to a methylene group has an activation barrier of 1.35 eV. The migration of the H atoms from the C-sites to the Mo-site to form a H2 molecule is an endothermic process. The adsorption energy for the second methane is −1.02 eV. The dissociation of the C–H bond of the methane has an activation barrier of 1.34 eV and a reaction energy of −0.59 eV. Reaction via the hydrocarbon pool mechanism is energetically favourable over this site. Activation of the methane C–H bond has a barrier of 1.18 eV and the methylation step is rather easy to occur with a barrier of 0.58 eV.
To compare the reactivity differences of Mo2C2 at different locations, all of the reaction energy profiles are summarized in Fig. S4–5 and Table S1 (ESI†). As can be seen from Fig. S4 (ESI†), the C–C bond coupling reaction step is the most difficult along the ethylene formation route and the reaction barrier over the β-site is the highest. The methylene group shows the highest stability over the γ-site compared to other locations, while the δ-site is the most favorable location for co-accommodation of the methylene and methyl groups and for stabilization of the C2H5 species. Fig. S5 (ESI†) shows that benzene methylation is the most difficult step and β- and γ-sites are not favorable for such a reaction. The reaction barriers over the α- and δ-sites are very close to each other, with the δ-site being preferable for both methylation and the subsequent deprotonation reactions.
Microkinetic modeling (MKM) was further carried out to evaluate the reactivity of different locations and to identify the rate-determining steps that govern the overall reaction rate. Fig. 3a shows the apparent activation barriers for the formation of ethylene from methane over different locations. This indicates that ethylene formation is favorable over the α-site with an apparent activation barrier (Eapp) of 0.65 eV. The Mo2C2 site located at all other locations is much less active with apparent activation barriers at least four-fold that over the α-site. The results of the degree of rate control (DRC) analysis are shown in Fig. S6 (ESI†). Very interestingly, it is found that the rate-determining step is also sensitive to the specific locations of the Mo2C2 active site in ZSM-5. The rate-determining step over the α-site is the initial C–H bond activation of methane, while the C–C bond coupling becomes dominant over the β- and δ-sites. In contrast, the reaction rate over the γ-site is controlled by the reaction steps related to molecular H2 formation.
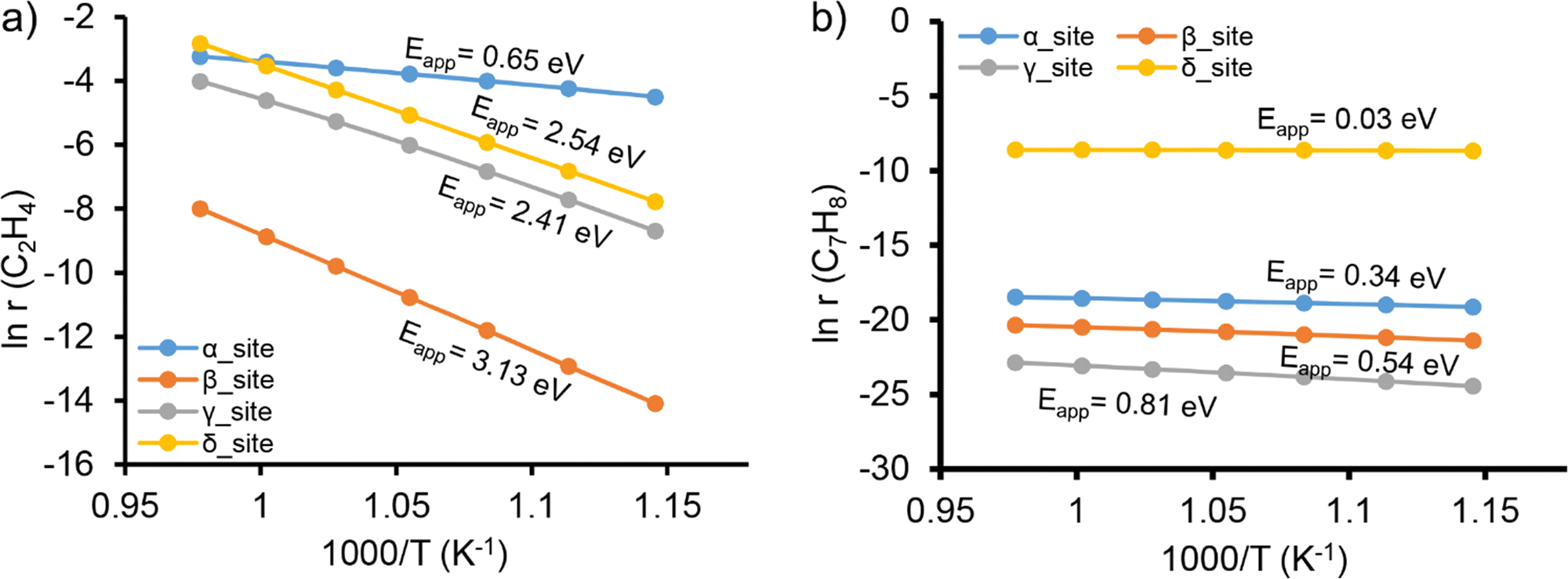 | ||
| Fig. 3 Microkinetic modeling of methane activation via (a) ethylene and (b) hydrocarbon pool pathways over the active site of [Mo2C2]2+ in ZSM-5. The formation rates r (in mol s−1) of ethylene as a function of temperature are presented. The apparent activation barriers (Eapp) indicated in the figures were calculated using the Arrhenius equation. Dual-site microkinetic models were considered. | ||
The MKM results of the hydrocarbon pool reaction pathway are illustrated in Fig. 3b and Fig. S7 (ESI†). Following this mechanism, it is identified that the δ-site is the most preferred location. For all sites evaluated, the apparent activation barriers along the hydrocarbon pool reaction pathway are much lower than the pathway of ethylene formation. Subsequent DRC analysis shows that benzene methylation to C7H9˙ is the main rate-determining step over the β-, δ-, and γ-sites, while over the α-site, the C7H9˙ dehydrogenation step turns out to have a more significant influence on the overall reaction rate. These results indicate the high degree of heterogeneity of MDA reactions taking place in Mo/ZSM-5.
In summary, the reactivity of Mo-carbide at different locations of the ZSM-5 matrix was evaluated by DFT calculations combined with microkinetic modeling. Distinct micro-environments can steer the reaction pathways and rate-determining steps. These results point out the complexity of MDA reaction and the significance of maximizing metal site utilization in the Mo/ZSM-5 catalyst by control of the Mo-carbide location.
This work was partially supported by Dr G. L.'s VENI grant (no. 016.Veni.172.034) awarded by Dutch Organization for Scientific Research (NWO). NWO-surfsara is acknowledged for providing access to the Dutch national e-infrastructure. The author thanks prof. E. A. Pidko (TUDelft) and Dr D. Sun (Supermicro) for fruitful discussions.
Conflicts of interest
There are no conflicts to declare.References
- O. V. Bragin, T. V. Vasina, A. V. Preobrazhenskii and K. M. Minachev, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1989, 38, 680 CrossRef.
- B. M. Weckhuysen, D. Wang, M. P. Rosynek and J. H. Lunsford, J. Catal., 1998, 175, 338 CrossRef CAS.
- (a) H. Cruchade, I. C. Medeiros-Costa, N. Nesterenko, J.-P. Gilson, L. Pinard, A. Beuque and S. Mintova, ACS Catal., 2022, 12, 14533 CrossRef CAS; (b) N. Kosinov, E. A. Uslamin, L. Meng, A. Parastaev, Y. Liu and E. J. M. Hensen, Angew. Chem., Int. Ed., 2019, 58, 7068 CrossRef CAS PubMed; (c) S. J. Han, S. K. Kim, A. Hwang, S. Kim, D.-Y. Hong, G. Kwak, K.-W. Jun and Y. T. Kim, Appl. Catal., B, 2019, 241, 305 CrossRef CAS.
- (a) W. Gao, Q. Wang, G. Qi, J. Liang, C. Wang, J. Xu and F. Deng, Angew. Chem., Int. Ed., 2023, e202306133 Search PubMed; (b) M. Çaǧlayan, A. L. Paioni, B. Dereli, G. Shterk, I. Hita, E. Abou-Hamad, A. Pustovarenko, A.-H. Emwas, A. Dikhtiarenko, P. Castaño, L. Cavallo, M. Baldus, A. D. Chowdhury and J. Gascon, ACS Catal., 2021, 11, 11671 CrossRef; (c) N. Kosinov and E. J. M. Hensen, Adv. Mater., 2020, 32, 2002565 CrossRef CAS PubMed.
- (a) I. Vollmer, G. Li, I. Yarulina, N. Kosinov, E. J. Hensen, K. Houben, D. Mance, M. Baldus, J. Gascon and F. Kapteijn, Catal. Sci. Technol., 2018, 8, 916 RSC; (b) J. Gao, Y. Zheng, J.-M. Jehng, Y. Tang, I. E. Wachs and S. G. Podkolzin, Science, 2015, 348, 686 CrossRef CAS PubMed.
- N. Kosinov, F. J. A. G. Coumans, G. Li, E. Uslamin, B. Mezari, A. S. G. Wijpkema, E. A. Pidko and E. J. M. Hensen, J. Catal., 2017, 346, 125 CrossRef CAS.
- (a) L. Liu, N. Wang, C. Zhu, X. Liu, Y. Zhu, P. Guo, L. Alfilfil, X. Dong, D. Zhang and Y. Han, Angew. Chem., Int. Ed., 2020, 59, 819 CrossRef CAS PubMed; (b) I. Vollmer, S. Ould-Chikh, A. Aguilar-Tapia, G. Li, E. Pidko, J.-L. Hazemann, F. Kapteijn and J. Gascon, J. Am. Chem. Soc., 2019, 141, 18814 CrossRef CAS PubMed; (c) M. Agote-Arán, A. B. Kroner, H. U. Islam, W. A. Sławiński, D. S. Wragg, I. Lezcano-González and A. M. Beale, ChemCatChem, 2019, 11, 473 CrossRef; (d) I. Lezcano-González, R. Oord, M. Rovezzi, P. Glatzel, S. W. Botchway, B. M. Weckhuysen and A. M. Beale, Angew. Chem., Int. Ed., 2016, 55, 5215 CrossRef PubMed.
- G. Li, I. Vollmer, C. Liu, J. Gascon and E. A. Pidko, ACS Catal., 2019, 9, 8731 CrossRef CAS.
- (a) W. Gao, G. Qi, Q. Wang, W. Wang, S. Li, I. Hung, Z. Gan, J. Xu and F. Deng, Angew. Chem., Int. Ed., 2021, 60, 10709 CrossRef CAS PubMed; (b) M. Çağlayan, A. Lucini Paioni, E. Abou-Hamad, G. Shterk, A. Pustovarenko, M. Baldus, A. D. Chowdhury and J. Gascon, Angew. Chem., Int. Ed., 2020, 59, 16741 CrossRef PubMed; (c) N. Kosinov, F. J. A. G. Coumans, E. A. Uslamin, A. S. G. Wijpkema, B. Mezari and E. J. M. Hensen, ACS Catal., 2017, 7, 520 CrossRef CAS.
- B. M. Weckhuysen, D. Wang, M. P. Rosynek and J. H. Lunsford, Angew. Chem., Int. Ed. Engl., 1997, 36, 2374 CrossRef CAS.
- N. Kosinov, A. S. G. Wijpkema, E. Uslamin, R. Rohling, F. J. A. G. Coumans, B. Mezari, A. Parastaev, A. S. Poryvaev, M. V. Fedin, E. A. Pidko and E. J. M. Hensen, Angew. Chem., Int. Ed., 2018, 57, 1016 CrossRef CAS PubMed.
- (a) N. Pfriem, P. H. Hintermeier, S. Eckstein, S. Kim, Q. Liu, H. Shi, L. Milakovic, Y. Liu, G. L. Haller, E. Baráth, Y. Liu and J. A. Lercher, Science, 2021, 372, 952 CrossRef CAS PubMed; (b) Z. Jin, L. Wang, E. Zuidema, K. Mondal, M. Zhang, J. Zhang, C. Wang, X. Meng, H. Yang, C. Mesters and F.-S. Xiao, Science, 2020, 367, 193 CrossRef CAS PubMed.
- (a) K. Cheng, L. C. J. Smulders, L. I. van der Wal, J. Oenema, J. D. Meeldijk, N. L. Visser, G. Sunley, T. Roberts, Z. Xu, E. Doskocil, H. Yoshida, Y. Zheng, J. Zečević, P. E. de Jongh and K. P. de Jong, Science, 2022, 377, 204 CrossRef CAS PubMed; (b) B. E. R. Snyder, P. Vanelderen, M. L. Bols, S. D. Hallaert, L. H. Böttger, L. Ungur, K. Pierloot, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Nature, 2016, 536, 317 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Method details, additional reaction energy diagrams and MKM results. See DOI: https://doi.org/10.1039/d3cc03517d |
| This journal is © The Royal Society of Chemistry 2023 |