
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst-in-class metallo-PROTAC as an effective degrader of select Pt-binding proteins†
Paul D.
O’Dowd
 ab,
Graeme P.
Sullivan
c,
Daniel A.
Rodrigues
a,
Tríona Ní
Chonghaile
c and
Darren M.
Griffith
*ab
ab,
Graeme P.
Sullivan
c,
Daniel A.
Rodrigues
a,
Tríona Ní
Chonghaile
c and
Darren M.
Griffith
*ab
aDepartment of Chemistry, Royal College of Surgeons in Ireland, Dublin 2, Ireland. E-mail: dgriffith@rcsi.ie
bSSPC, the Science Foundation Ireland Research Centre for Pharmaceuticals, Ireland
cDepartment of Physiology and Medical Physics, Royal College of Surgeons in Ireland, Dublin 2, Ireland
First published on 28th September 2023
Abstract
We report the development of the first metallo-PROTAC, specifically a Pt-PROTAC, that can effectively degrade select Pt(II)-binding proteins. The Pt-PROTAC prototype successfully degraded thioredoxin-1 and thioredoxin reductase-1 in multiple myeloma cancer cell lines. Metallo-PROTACs will have important applications in the identification of metal binding proteins and as chemotherapeutic agents.
Metal-based drugs play important clinical roles as therapeutic agents. Metal–protein interactions are central to the mechanisms of action of many of these drugs, though much remains to be discovered.1,2 Platinum (Pt)-based drugs are the most well-known class of metal-based drugs and are employed in nearly 50% of all anticancer chemotherapeutic regimens.3,4 Three Pt(II) drugs, cisplatin, carboplatin, and oxaliplatin are approved worldwide for clinical use. Together, they are used to treat numerous different cancer types including head and neck, gynaecological, respiratory, breast cancers, upper gastrointestinal, urogenital, colorectal, lymphomas, sarcomas, and multiple myelomas.5
Traditionally the mechanism of action of Pt(II) drugs has been primarily linked to DNA adduct formation, as the electrophilic Pt(II) centre readily binds DNA bases. The resulting monofunctional Pt–DNA adducts and DNA inter- and intrastrand crosslinks impair DNA function and induce cellular apoptosis.6,7 Remarkably only 1% of intracellular cisplatin binds nuclear DNA and Pt drugs exert noteworthy cytotoxic effects in enucleated cells. Pt(II) anticancer drugs also react with a range of other nucleophiles, including RNA bases, mitochondrial DNA and proteins.7,8 It is also noteworthy that oxaliplatin can induce immunogenic cell death (ICD)9–11 and target ribosome biogenesis resulting in the activation of nucleolar stress response pathways.12,13 There has been recent increased interest in the effects of Pt protein binding and in particular the role that Pt protein binding plays in on- and off-target activity of Pt-based drugs.14–18
It's well known that Pt(II) as a soft centre (HSAB) has an affinity for the S atoms in the amino acids cysteine and methionine and readily binds the cysteine containing tripeptide, glutathione, and the cysteine rich metallotheionin. Pt(II) also coordinates to histidine via the more borderline N atoms of imidazole as well as to many other amino acids (e.g. tyrosine (Y), serine (S), glutamic acid (E), aspartic acid (D) and lysine (K)).16 In turn Pt anticancer drugs have been shown to bind to a multitude of blood plasma16 and cellular proteins.14,17,19 Recently for example, azidoplatin, a cisplatin mimic which possesses an azide handle, was developed and employed by DeRose and coworkers in a novel post treatment click-based chemical proteomic method to label and isolate platinated proteins in S. cerevisiae. Significantly 152 Pt(II)-bound proteins were identified including several proteins implicated in the endoplasmic reticulum stress response.20 Certainly, novel techniques that knockdown or silence proteins should further elucidate unknown metal–protein interactions.14
Proteolysis targeting chimeras (PROTACs) have recently emerged as a technology that can efficiently degrade target proteins.21–24 PROTACs are bifunctional molecules that hijack the ubiquitin proteasome system (UPS) to achieve degradation of proteins of interest (POIs) such as disease-related target proteins. Significantly the selective degradation of a broad spectrum of protein targets from transcription factors to enzymes has been reported. PROTACs consist of a ligand that binds to an E3 ligase, connected by an appropriately designed linker to a second ligand that binds to particular POIs, Fig. 1. A functional PROTAC instigates the formation of a ternary complex, POI-PROTAC-E3 ligase. PROTACs therefore recruit E3 ligases to the vicinity of the POI, promoting its ubiquitination and subsequent degradation by the proteasome through proteolysis.20,25–27 Thus, successful degradation relies on an adequate affinity of the PROTAC toward both the E3 ligase and the POI.6 Significantly PROTACs exhibit catalytic behaviour and can induce proteasomal degradation at substoichiometric levels.21
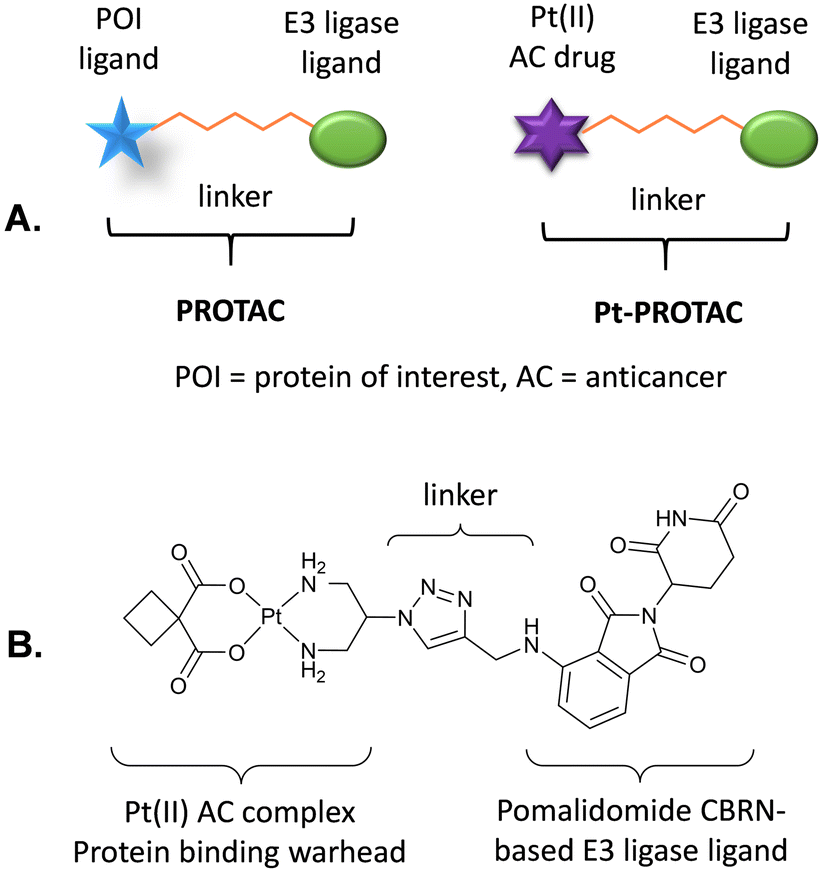 | ||
| Fig. 1 (A) Generalised structures of a PROTAC and a Pt-PROTAC. (B) Structure of novel Pt-PROTAC reported in this study. | ||
A number of well-known E3 ligase ligands have been successfully employed in PROTACs including cereblon (CRBN), Von Hippel Lindau (VHL), mouse double minute 2 homolog (MDM2) and cellular inhibitor of apoptosis protein 1 (cIAP1).22 Of these, based on ligand selection, CRBN is the most commonly recruited E3 ligase. This is typically achieved using thalidomide and derivatives, such as pomalidomide, as the CRBN E3 ligase ligands in PROTACs.27,28 CRBN-based PROTACs have been developed to target a diverse range of proteins of interest, including the bromodomain and extra-Terminal (BET) proteins (BRD2/3/4), FKBP12, BCR-ABL, BRD9, Sirt2, CDK9, FLT3, BTK, ALK, CDK4/CDK6 and HDAC6.21 Furthermore ARV-471, a PROTAC estrogen receptor degrader, has entered Phase III clinical trials for treatment of advanced breast cancer.29 Thus, we hypothesised that the design of a first-in-class metallo-PROTAC could lead to efficient degradation of metal binding proteins via the UPS.
With this goal in mind, we set out to develop a Pt-PROTAC as the first metallo-PROTAC and demonstrate that this Pt-PROTAC can effectively degrade Pt binding proteins (PtBPs) via the UPS. This novel Pt-PROTAC consists of an E3 ligase ligand connected by a linker to a Pt(II) centre via a stable amine carrier ligand and where the Pt(II) centre acts as a protein targeting warhead. Protein binding by this Pt-PROTAC will therefore instigate the formation of a ternary complex, PtBP-PtPROTAC-E3 ligase, and recruit E3 ligases to the vicinity of the PtBP, promoting ubiquitination and subsequent degradation by the proteasome through proteolysis.21,25–27
As a proof of concept study we designed a novel Pt-PROTAC prototype, consisting of pomalidomide as the CRBN-based E3 ligase ligand, connected by a short triazole-based linker to a carboplatin-like Pt(II) complex as the protein targeting warhead, Fig. 1. We also designed a deactivated Pt-PROTAC (D-Pt-PROTAC) as a negative control, in which the pomalidomide E3 ligase ligand is N-methylated rendering it incapable of binding to the E3 ligase.
Pt-PROTAC and deactivated Pt-PROTAC control compound (D-Pt-PROTAC) were synthesised by conjugating cis-[Pt(2- azidopropane-1,3-diamine)(CBDCA-2H)] ([Pt(DAP-N3)(CBDCA-2H)]) 1 with CRBN E3 ligand pomalidomide 2 or deactivated CRBN E3 ligand pomalidomide 3 through a copper-catalysed azide–alkyne cycloaddition (CuAAC) reaction. These complexes were fully characterised by 1H and 13C NMR and HRMS and purities of >95% were verified by RP-HPLC, Fig. S1 to S15 (ESI†).
We subsequently investigated whether Pt-PROTAC could degrade thioredoxin-1 (TRX1), thioredoxin reductase-1 (TRX-R1) and glutathione S transferase (GTSP1), all three of which are known to be bound by Pt(II) centres.30–35 We rationalised, with reference to protein X-ray crystal structures, that our Pt-PROTAC prototype would be more likely to degrade TRX1 and TRX-R1 as opposed to GTSP1, due to the positioning of binding sites on the respective proteins. In order to investigate the capability of Pt-PROTAC to degrade these proteins, multiple myeloma cell lines (JJN3 and MM1.S) were incubated with either 100 or 150 μM of the complex for 24 hours, and the degradation subsequently investigated by western blot analysis.
As per Fig. 2 and Fig. S16 (ESI†), Pt-PROTAC demonstrated pronounced degradation of TRX1 and TRX-R1 in both cell lines whereas GTSP1 was not markedly degraded. Importantly, carboplatin and pomalidomide failed to degrade any of the three proteins investigated in a control experiment in JJN3 cells, Fig. 2. The control D-Pt-PROTAC, which possesses an N-methylated glutarimide moiety, also exhibited no degradation activity for all three proteins. Furthermore pretreatment of cell lines with proteasome inhibitor, bortezomib (1 nM) for 30 min, resulted in a failure of Pt-PROTAC target degradation, thereby implicating the UPS in the observed degradation of TRX1 and TRX-R1 by Pt-PROTAC, Fig. 2 and Fig. S16 (ESI†).
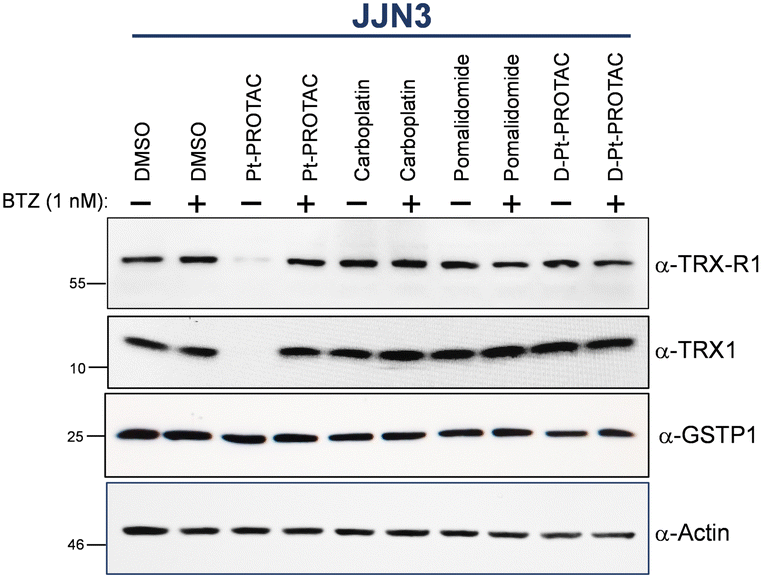 | ||
| Fig. 2 Western blot analysis of whole cell lysates of JJN3 cells after treatment with equimolar concentrations (150 μM) of Pt-PROTAC, carboplatin, pomalidomide and D-Pt-PROTAC as well as vehicle control for 24 h with (+) or without (−) the presence of the proteasome inhibitor, bortezomib (1 nM). | ||
PROTAC linker length is a key determinant of degradation efficiency, with longer linkers typically more successful at degrading proteins with deep binding pockets.36 Pt(II)-binding sites have previously been reported for TRX1,30,37 TRX-R133 and GTSP1.35 We hypothesise therefore that our Pt-PROTAC, which possesses a short triazole based linker, can readily access and bind to key amino acids residues in TRX1 and TRX-R1 though not in GTSP1.
C32 and C35 in TRX1 and C498 in TRX-R1 are solvent exposed and near the surface of the respective proteins, Fig. 3(A) and (B).30,33 Binding of Pt-PROTAC at close proximity to the proteins’ surfaces thus allows successful E3 ligase recruitment and subsequent UPS mediated degradation. GTSP1 exists as a dimer within mammalian cells and incubation with Pt has been shown to bridge this dimer interface.38 This bridging occurs through Pt-interactions with cysteines C47 and more significantly C101 within the protein, Fig. 3(C).35 Importantly, these binding sites are positioned towards the centre of the protein and thus effective protein degradation is likely being inhibited through impaired Pt-protein binding or unsuccessful E3 ligase recruitment.
 | ||
| Fig. 3 (A) X-ray structure of the human thioredoxin (PDB ID: 1ERT) highlighting the cysteines residues C32 and C35. (B) X-ray structure of human thioredoxin reductase I and a terpyridine platinum(II) complex (PDB ID: 2ZZB) highlighting the bond between Pt(II) and the cysteine 498 and the stacking of the terpyridine with tryptophan 114 in a solvent-exposed region. (C) X-ray structure of human glutathione S-transferase in complex with cisplatin in the presence of glutathione (PDB ID: 5DJL) highlighting that Pt(II) is binding in a more deep pocket of the protein. | ||
Cell death analysis was investigated by annexin V/PI staining using flow cytometry. IC50s were determined for carboplatin and Pt-PROTAC against JJN3 and MM1.S cell lines, Table 1 and Fig. S17 and S18 (ESI†). From these experiments, we found Pt-PROTAC to be approximately 3-fold less effective at killing these cell lines in comparison to carboplatin.
| Complex | JJN3 | MM1.S |
|---|---|---|
| Carboplatin | 49.37 | 47.22 |
| Pt-PROTAC | 154.1 | 172.2 |
| D-Pt-PROTAC | >500 | >500 |
It is noteworthy that PROTACs can suffer from relatively poor cell permeability, which may be a factor in the higher IC50 values observed for Pt-PROTAC as compared to carboplatin.39 JJN3 cells were also treated with carboplatin, Pt-PROTAC and D-Pt-PROTAC at concentrations ranging from 10 to 200 μM and at 24 and 48 hours, Fig. S19 (ESI†). The vast majority of cell death appears to occur within 24 hours post-treatment at 200 μM with modest differences in the efficacy of carboplatin and Pt-PROTAC. D-Pt-PROTAC, on the other hand caused minimal cell death across all concentrations tested. Ultimately Pt-PROTAC was found to be relatively non-cytotoxic in comparison to carboplatin.
In this proof of concept study, a novel Pt-PROTAC was successfully developed as an effective degrader of Pt(II)-binding proteins, TRX1 and TRX-R1 via the UPS. Furthermore this complex is relatively non-cytotoxic and kills cancer cells to a significantly lower extent than carboplatin as evidenced by flow cytometry results. Metallo-PROTACs offer much potential through rational design of complexes for the selective degradation of metal-binding proteins. Protein selectivity can be fine-tuned via the (i) optimization of linker type and length, (ii) selection of E3 ligase ligand and (iii) selection of metal-based protein binding warhead. Ultimately we believe in the coming years metallo-PROTACs will play a central role (i) in the identification of metal binding proteins via a novel metalloproteomic technique and (ii) as chemotherapeutic agents.
Paul O’Dowd: conceptualisation, methodology, investigation, writing – review and editing, Daniel Alencar: conceptualisation, methodology, investigation, writing – review and editing. Graeme Sullivan: methodology, investigation, writing – review and editing, Triona Ní Chonghaile: conceptualisation, resources, supervision, methodology, writing – review and editing, funding acquisition, Darren M. Griffith: conceptualisation, resources, methodology, writing – original draft preparation, supervision, project administration, funding acquisition.
DMG and POD gratefully acknowledge funding received from the Synthesis and Solid State Pharmaceutical Centre (SSPC), financed by a research grant from Science Foundation Ireland (SFI) and co-funded under the European Regional Development Fund under Grant Number 12/RC/2275_P2. TNC gratefully acknowledges the Irish Research Council (IRCLA/2022/2822) and SFI (19/FFP/646) for funding. DAR gratefully acknowledges the Irish Research Council (GOIPD/2022/764) for funding. GPS thanks the Irish Cancer Society under grant number CRF21SUL for funding.
Conflicts of interest
There are no conflicts to declare.References
- T. Marzo, G. Ferraro, A. Merlino and L. Messori, Encycl. Inorg. Bioinorg. Chem., 2020, 1–17, DOI:10.1002/9781119951438.eibc2747
.
- L. Skos, Y. Borutzki, C. Gerner and S. M. Meier-Menches, Curr. Opin. Chem. Biol., 2023, 73, 102257 CrossRef CAS PubMed
- S. Alassadi, M. J. Pisani and N. J. Wheate, Dalton Trans., 2022, 51, 10835–10846 RSC
- E. Armstrong-Gordon, D. Gnjidic, A. J. McLachlan, B. Hosseini, A. Grant, P. J. Beale and N. J. Wheate, J. Cancer Res. Clin. Oncol., 2018, 144, 1561–1568 CrossRef PubMed
- I. S. Um, E. Armstrong-Gordon, Y. E. Moussa, D. Gnjidic and N. J. Wheate, Inorg. Chim. Acta, 2019, 492, 177–181 CrossRef CAS
- S. Rottenberg, C. Disler and P. Perego, Nat. Rev. Cancer, 2021, 21, 37–50 CrossRef CAS PubMed
- I. A. Riddell, Met. Ions Life Sci., 2018, 18, 1–42 CrossRef CAS PubMed
- L. Galluzzi, I. Vitale, J. Michels, C. Brenner, G. Szabadkai, A. Harel-Bellan, M. Castedo and G. Kroemer, Cell Death Dis., 2014, 5, e1257 CrossRef CAS PubMed
- B. Englinger, C. Pirker, P. Heffeter, A. Terenzi, C. R. Kowol, B. K. Keppler and W. Berger, Chem. Rev., 2019, 119, 1519–1624 CrossRef CAS PubMed
- S. Göschl, E. Schreiber-Brynzak, V. Pichler, K. Cseh, P. Heffeter, U. Jungwirth, M. A. Jakupec, W. Berger and B. K. Keppler, Metallomics, 2017, 9, 309–322 CrossRef PubMed
- A. Tesniere, F. Schlemmer, V. Boige, O. Kepp, I. Martins, F. Ghiringhelli, L. Aymeric, M. Michaud, L. Apetoh, L. Barault, J. Mendiboure, J. P. Pignon, V. Jooste, P. van Endert, M. Ducreux, L. Zitvogel, F. Piard and G. Kroemer, Oncogene, 2010, 29, 482–491 CrossRef CAS PubMed
- P. M. Bruno, Y. Liu, G. Y. Park, J. Murai, C. E. Koch, T. J. Eisen, J. R. Pritchard, Y. Pommier, S. J. Lippard and M. T. Hemann, Nat. Med., 2017, 23, 461–471 CrossRef CAS PubMed
- P. D. O'Dowd, D. F. Sutcliffe and D. M. Griffith, Coord. Chem. Rev., 2023, 497, 215439 CrossRef
- E. C. Sutton, C. E. McDevitt, M. V. Yglesias, R. M. Cunningham and V. J. DeRose, Inorg. Chim. Acta, 2019, 498, 118984 CrossRef CAS
- N. J. Farrer and D. M. Griffith, Curr. Opin. Chem. Biol., 2020, 55, 59–68 CrossRef CAS PubMed
- J. Wang, J. Tao, S. Jia, M. Wang, H. Jiang and Z. Du, Pharmaceuticals, 2021, 14 Search PubMed
- O. Pinato, C. Musetti and C. Sissi, Metallomics, 2014, 6, 380–395 CrossRef CAS PubMed
- X. Wang, Y. Zhang and C. Wang, RSC Chem. Biol., 2023, 4, 670–674 RSC
- C. Bischin, A. Lupan, V. Taciuc and R. Silaghi-Dumitrescu, Mini Rev. Med. Chem., 2011, 11, 214–224 CrossRef CAS PubMed
- R. M. Cunningham and V. J. DeRose, ACS Chem. Biol., 2017, 12, 2737–2745 CrossRef CAS PubMed
- M. Konstantinidou, J. Li, B. Zhang, Z. Wang, S. Shaabani, F. Ter Brake, K. Essa and A. Dömling, Expert Opin. Drug Discovery, 2019, 14, 1255–1268 CrossRef CAS PubMed
- M. Békés, D. R. Langley and C. M. Crews, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef PubMed
- S. Gu, D. Cui, X. Chen, X. Xiong and Y. Zhao, BioEssays, 2018, 40, e1700247 CrossRef PubMed
- M. He, C. Cao, Z. Ni, Y. Liu, P. Song, S. Hao, Y. He, X. Sun and Y. Rao, Signal Transduction Targeted Ther., 2022, 7, 181 CrossRef CAS PubMed
- M. G. Jaeger and G. E. Winter, Cell Chem. Biol., 2020, 27, 14–16 CrossRef CAS PubMed
- D. Alencar Rodrigues, A. Roe, D. Griffith and T. Chonghaile, Curr. Top. Med. Chem., 2022, 22, 408–424 CrossRef PubMed
- A. Bricelj, C. Steinebach, R. Kuchta, M. Gütschow and I. Sosič, Front. Chem., 2021, 9 Search PubMed
- T. Ito, H. Ando, T. Suzuki, T. Ogura, K. Hotta, Y. Imamura, Y. Yamaguchi and H. Handa, Science, 2010, 327, 1345–1350 CrossRef CAS PubMed
- C. X. Ma, M. D. Laurentiis, H. Iwata, S. A. Hurvitz, S. A. Wander, M. A. Danso, D. R. Lu, J. P. Smith, Y. Liu, L. Tran, S. Anderson and E. P. Hamilton, J. Clin. Oncol., 2023, 41, TPS1122 CrossRef
- M. Kato, H. Yamamoto, T.-A. Okamura, N. Maoka, R. Masui, S. Kuramitsu and N. Ueyama, Dalton Trans., 2005, 1023–1026, 10.1039/B419119F
- E. S. J. Arnér, H. Nakamura, T. Sasada, J. Yodoi, A. Holmgren and G. Spyrou, Free Radical Biol. Med., 2001, 31, 1170–1178 CrossRef PubMed
- J. Du, Y. Wei, Y. Zhao, F. Xu, Y. Wang, W. Zheng, Q. Luo, M. Wang and F. Wang, Inorg. Chem., 2018, 57, 5575–5584 CrossRef CAS PubMed
- Y.-C. Lo, T.-P. Ko, W.-C. Su, T.-L. Su and A. H. J. Wang, J. Inorg. Biochem., 2009, 103, 1082–1092 CrossRef CAS PubMed
- A.-B. Witte, K. Anestål, E. Jerremalm, H. Ehrsson and E. S. J. Arnér, Free Radical Biol. Med., 2005, 39, 696–703 CrossRef CAS PubMed
- A. De Luca, L. J. Parker, W. H. Ang, C. Rodolfo, V. Gabbarini, N. C. Hancock, F. Palone, A. P. Mazzetti, L. Menin, C. J. Morton, M. W. Parker, M. Lo Bello and P. J. Dyson, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13943–13951 CrossRef CAS PubMed
- R. I. Troup, C. Fallan and M. G. J. Baud, Explor. Target Antitumor. Ther., 2020, 1, 273–312 Search PubMed
- J. Will, W. S. Sheldrick and D. Wolters, J. Biol. Inorg. Chem., 2008, 13, 421–434 CrossRef CAS PubMed
- W. Harshbarger, S. Gondi, S. B. Ficarro, J. Hunter, D. Udayakumar, D. Gurbani, W. D. Singer, Y. Liu, L. Li, J. A. Marto and K. D. Westover, J. Biol. Chem., 2017, 292, 112–120 CrossRef CAS PubMed
- C. Pu, S. Wang, L. Liu, Z. Feng, H. Zhang, Q. Gong, Y. Sun, Y. Guo and R. Li, Chin. Chem. Lett., 2023, 34, 107927 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc03340f |
| This journal is © The Royal Society of Chemistry 2023 |