
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced anion recognition by ammonium [2]catenane functionalisation of a halogen bonding acyclic receptor†
Thanthapatra
Bunchuay
 ab,
Theerapat
Khianjinda
b,
Pasit
Srisawat
b,
Yuen Cheong
Tse
a,
Christian
Gateley
a and
Paul D.
Beer
*a
ab,
Theerapat
Khianjinda
b,
Pasit
Srisawat
b,
Yuen Cheong
Tse
a,
Christian
Gateley
a and
Paul D.
Beer
*a
aDepartment of Chemistry, Chemistry Research Laboratory, University of Oxford, Mansfield Road, Oxford OX1 3TA, UK. E-mail: paul.beer@chem.ox.ac.uk
bDepartment of Chemistry and Center for Excellence for Innovation in Chemistry (PERCH-CIC), Faculty of Science, Mahidol University, Bangkok, 10400, Thailand
First published on 24th October 2023
Abstract
Ammonium-dibenzo[24]crown-8 [2]catenane functionalisation of a 3,5-bis-iodotriazole-pyridine motif produces a potent halogen bonding (XB) receptor capable of binding anions in aqueous-acetone solvent mixtures of up to 20% water. Exploiting the kinetically inert nature of the mechanically bonded cationic ammonium [2]catenane substituents, the XB receptor is demonstrated to exhibit superior anion recognition behaviour in comparison to labile sodium cation complexed bis-benzo[15]crown-5 XB and HB triazole-pyridine heteroditopic receptor analogues.
The continued interest in developing host systems for the molecular recognition of charged species is in no small part due to the pervasiveness of ions in a multitude of fundamental and essential chemical and biological processes.1,2 The characteristics of counterions have a significant impact on the cation and anion binding affinities of monotopic receptor systems. Consequently, the development of heteroditopic receptors for ion-pair recognition, where favourable proximal cation–anion electrostatic interactions and conformational allosteric cooperativity boost charged guest binding strength, has gained increasing attention in supramolecular host–guest chemistry research for the past few decades.3 Notably, heteroditopic hosts have demonstrated a plethora of potential applications including the extraction and recovery of transition and main-group metals, the solubilisation of inorganic salts in organic media, membrane transport and recognition of biologically-relevant zwitterions.4–6
Ion-pair recognition is associated with multiple complex equilibria. Strong ion-pairing, particularly in low polarity organic solvents, inhibits the complete dissociation of charged species, and only a fraction of free ions persists in solution.7 With heteroditopic receptors, the cation and anion are bound simultaneously, taking advantage of cooperativity between the proximal respective recognition sites. Commonly employed cation binding motifs in heteroditopic host design are crown ethers, and to achieve anion binding, hydrogen bond (HB) donors such as amide, urea and triazole are frequently used.8–11 During the last decade, the emergence of sigma–hole interactions such as halogen bonding (XB) has demonstrated, in general, their superior anion guest binding capabilities compared to hydrogen bonding (HB) in host design.12–15
Recently, we reported a series of heteroditopic receptors containing XB donor groups covalently linked to benzo[15]crown-5 motifs, wherein co-bound sodium cation crown ether complexation cooperatively augmented bromide and iodide binding strength.16 Herein, we present an alternative novel post-mechanical bond synthesis strategy, where a positively charged ammonium [2]catenane motif replaces the crown ether complexed alkali metal cation. Stoddart's pioneering work demonstrating pseudorotaxane assembly between dibenzo[24]crown-8 and the dibenzyl ammonium group, stabilised by strong ion-dipole HB interactions,17 has led to its extensive employment in the mechanical interlocked molecule (MIM) synthesis of an enormous range of rotaxane and catenane supramolecular architectures.18–20
A mechanically bonded dibenzo[24]crown-8 dibenzylammonium [2]catenane synthon was employed in the construction of a novel XB 3,5-bis-iodotriazole [2]catenane functionalised pyridyl receptor 1·(PF6)2 for anion recognition in aqueous media (Fig. 1a). Extensive 1H-NMR anion titration investigations demonstrate the superior anion recognition properties of 1·(PF6)2 in comparison to bis-sodium cation complexed benzo[15]crown-5 functionalised XB and HB heteroditopic receptor analogues (XB9·2NaPF6 and HB10·2NaPF6, respectively). These observations serve to highlight the importance of the kinetically inert nature of the cationic [2]catenane group in enhancing anion binding in competitive aqueous media, as an alternative to a kinetically labile alkali metal cation complexed crown ether.
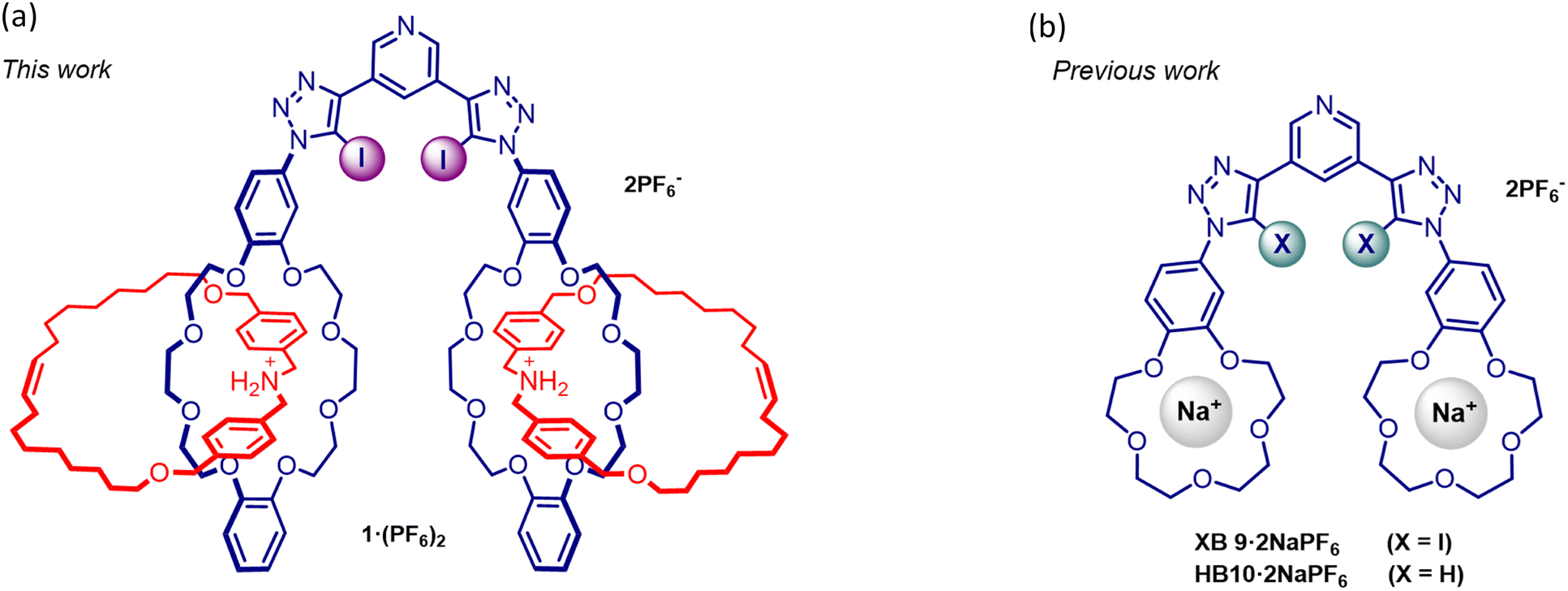 | ||
| Fig. 1 Chemical structures of (a) ammonium-dibenzo[24]crown-8 [2]catenane functionalised XB receptor, 1·(PF6)2 and (b) XB and HB heteroditopic benzo-15-crown appended analogous receptors containing sodium ion complexed benzo[15]crown-5, XB9·2NaPF6 and HB10·2NaPF6, respectively. | ||
In order to prepare azide functionalised ammonium-crown ether [2]catenane synthon 8·PF6 for subsequent CuAAC click XB reaction, the initial syntheses of azido-dibenzo[24]crown-8 5 and the appropriate bis-alkene ammonium thread 6·PF6,21–23 were undertaken using modified procedures (Scheme S1, ESI†). To prepare the macrocycle 5, the bis-tosylate precursor 2 was first prepared from an alkylation reaction of catechol, followed by tosylation. Subsequent macrocyclization was achieved via a potassium cation template ring closing reaction using a modified literature procedure.24 An equimolar solution of the bis-tosylate 2 and 2-nitrocatechol in dry MeCN was refluxed in the presence of KPF6 and K2CO3 to give nitro-functionalised dibenzo[24]crown-8 3 in 87% yield. Reduction of 3 followed by diazotization and a nucleophilic aromatic substitution reaction with NaN3 gave azido-dibenzo[24]crown-8 5 in 90% yield (Scheme 1).
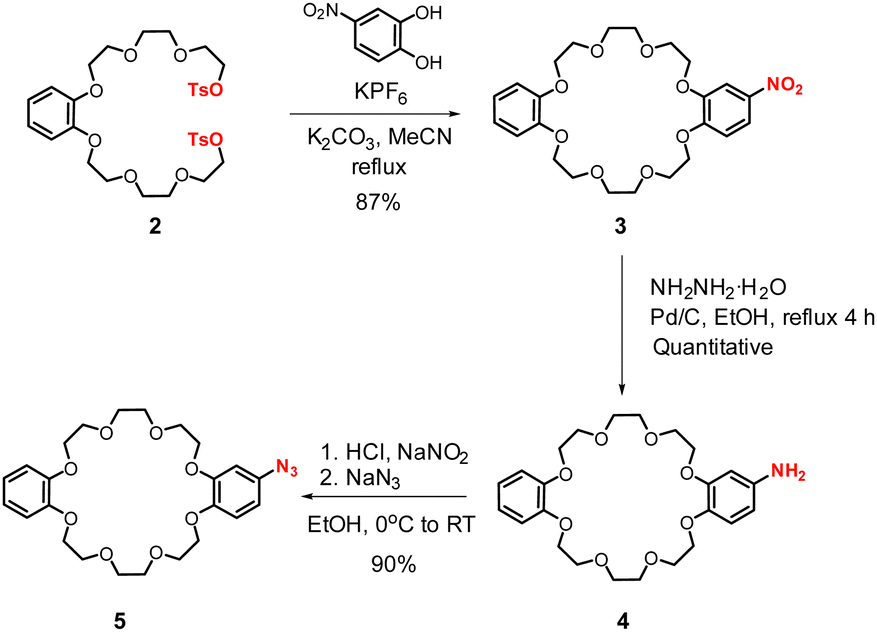 | ||
| Scheme 1 Synthesis of azide functionalised dibenzo[24]crown-8, 5. | ||
Qualitative 1H-NMR spectroscopic studies of pseudorotaxane-complex formation between thread 6·PF6 and macrocycle 5 were conducted by mixing an equimolar solution of each component in d2-dichloromethane at room temperature. The 1H-NMR spectrum of the resulting homogeneous solution showed well resolved signals of both complexed and uncomplexed states, indicating the pseudorotaxane equilibrium was in slow exchange on the NMR timescale (Fig. S17, ESI†). The aromatic proton signals from the threading component 6·PF6 (Hc and Hb) displayed an upfield shift upon interpenetration, concomitantly with the observation of substantial chemical shift changes of proton signals associated with the macrocyclic component. These evidences all indicate the formation of pseudorotaxane 7·PF6, in which hydrogen bonding and ammonium cation–dipole (NH+–O) interactions, along with π–π interactions stabilise the complexed assembly in d2-dichloromethane.
The stability of the pseudorotaxane complex assembly allowed for the preparation of the azide appended [2]catenane 8·PF6via a ring closing metathesis reaction using Grubbs’ 1st-generation catalyst.25,26 The crude product was purified by preparative silica gel thin-layer chromatography, followed by size exclusion chromatography and anion exchange to afford the azido [2]catenane 8·PF6 product in 30% yield (Scheme 2). The interlocked nature of catenane 8·PF6 was confirmed by 1H–1H ROESY NMR spectroscopy (Fig. S12, ESI†).
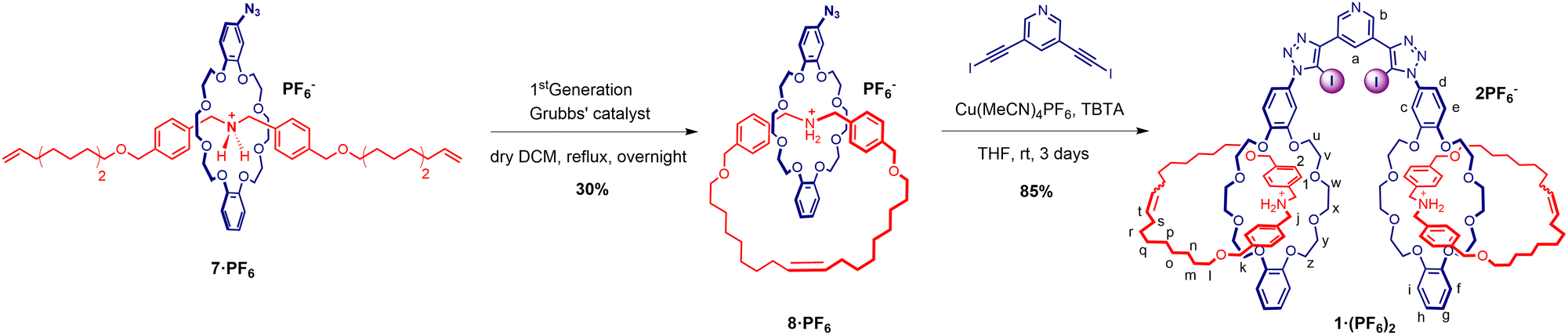 | ||
| Scheme 2 Synthetic scheme for azide functionalised [2]catenane 8·PF6via a ring closing metathesis and [2]catenane functionalised XB receptor 1·(PF6)2. | ||
The CuAAC reaction of two equivalents of 8·PF6 with 3,5-bis-iodoethylnylpyridine27 gave the target product 1·(PF6)2 in an excellent 85% yield after purification (Scheme 2). The 1H-NMR spectrum of 1·(PF6)2 shows broad proton signals from the pyridyl ring (Ha and Hb) hidden under signals of aromatic crown ether protons with a significant downfield perturbation of aryl protons (Hc, Hd, and He) compared to those in the starting material 8·PF6 (Fig. 2a). Additionally, the methylene proton (Hj, Hk, and Hu–z) resonances of 1·(PF6)2 are split into multiple signals whilst the alkyl proton (Hl–s) resonances remain unchanged. Moreover, the HRMS of 1·(PF6)2 revealed a signal of m/z = 1185.02467, corresponding to [M-2PF6−]2+, in agreement with the theoretical mass spectrum (Fig. 2b and ESI†).
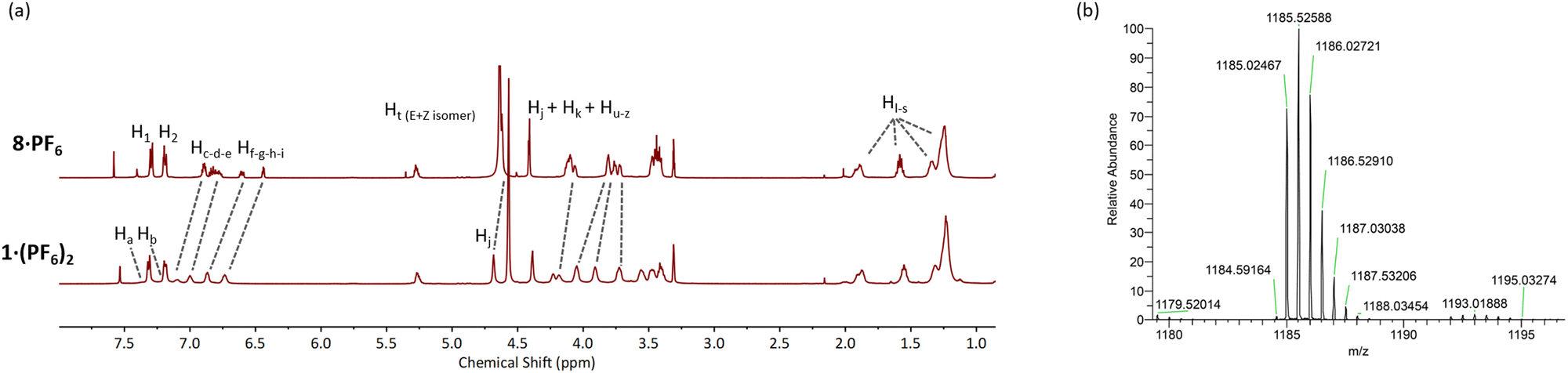 | ||
Fig. 2 (a) Comparative truncated 1H-NMR spectra of 8·PF6 and 1·(PF6)2 in CD3OD/CDCl3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) and (b) HRMS of 1·(PF6)2. :1 v/v) and (b) HRMS of 1·(PF6)2. | ||
The anion recognition properties of 1·(PF6)2 were initially investigated by 1H-NMR spectroscopic titration experiments in the organic solvent mixture 10% d6-DMSO/CDCl3. Upon addition of iodide as tetrabutylammonium iodide (TBAI), the chemical shift of the internal pyridyl proton (Ha) displayed downfield perturbations as a function of iodide anion concentration. WinEQNMR2 analysis28 of the titration data determined a 1:1 host–guest stoichiometric association constant (Ka) of >104 M−1, while the sodium cation complexed XB receptor XB9·2NaPF6 reported previously exhibited an apparent Ka of 5660 M−1 for iodide binding in this solvent system.16 Importantly this initial observation suggested appending a kinetically stable ammonium-crown ether [2]catenane substituent to the XB anion recognition motif significantly enhanced iodide binding affinity in comparison to XB9·2NaPF6.
Anion binding studies were further undertaken in the more competitive aqueous-organic solvent mixture 5% D2O/d6-acetone. Addition of TBAI to 1·(PF6)2 resulted in a large downfield perturbation (Δδ = +0.2 ppm, 10 equiv. I−) of the internal pyridyl proton signal (Ha) with a concomitant splitting of the resonances arising from the phenyl protons (Hc, Hd, He), linked to the iodotriazole motifs. In contrast, the crown ether protons (Hu–z) and the methylene protons adjacent to the ammonium cation group (Hj) showed negligible perturbations, clearly indicating that the halide anion was bound in the XB cleft of the receptor and not interacting directly with the ammonium–crown ether catenane complex (Fig. S18, ESI†).
Although initially only sparingly soluble in the 5% D2O/d6-acetone solvent mixture, the addition of two equivalents of NaPF6 resulted in the dissolution of both XB and HB heteroditopic receptors, indicating respective sodium cation-benzo[15]crown-5 complexation. However, it should be noted that under these experimental conditions, metal cation binding is not quantitative and there remains at equilibrium a significant proportion of un-complexed metal cation and heteroditopic receptor. It is noteworthy that this is not the case with the kinetically inert cationic [2]catenane motif in 1·(PF6)2. WinEQNMR228 and Bindfit analysis29 of the anion titration data monitoring the shifts of the internal pyridyl proton (Ha) determined 1:1 stoichiometric anion-host association constants together with apparent association constant data obtained from analogous titrations with XB9·2NaPF6 and HB10·2NaPF6 (Table 1). Notably, the catenane functionalised XB receptor 1·(PF6)2 bound the heavier halides much more strongly than either XB9·2NaPF6 or HB10·2NaPF6, displaying at least over ca. 20-fold and ca. 40-fold enhancement of iodide and bromide binding strength respectively. Impressively, 1·(PF6)2 also displayed binding of highly hydrated anions such as Cl− and SO42− with Ka values of 44 and 293 M−1 respectively. In 20% D2O/d6-acetone, the maximum amount of water content where the host maintained sufficient solubility for 1H-NMR spectroscopic titration, 1·(PF6)2 was still able to bind iodide strongly (Ka = 860 M−1). This observation clearly demonstrates that the combination of mechanically interlocked ammonium cation dibenzo[24]crown-8 containing substituents appended to a XB anion binding scaffold is as an effective novel methodology to achieve anion recognition in highly competitive aqueous solvent mixtures. It is also noteworthy that the superbase property of such ammonium MIM substituents potentially enables the positive charge to be retained even under basic aqueous solvent medium conditions.30
| Anion | Association constant (Ka, M−1) | ||
|---|---|---|---|
| 1·(PF6)2 | XB9·2NaPF6 | HB10·2NaPF6 | |
|
a
K
a values obtained by fitting the binding isotherm (monitored chemical shift changes of Ha) to a host–guest 1:1 stoichiometric binding model using WinEQNMR2; error(±) is less than 10%; each anion added as its tetrabutylammonium (TBA) salt.
b Titrations in the presence of 2 equivalents of sodium cations added as NaPF6.
c Titration in 20% D2O/d6-acetone.
d Fitting with Bindfit v.05.29
|
|||
| Cl− | 44 | Precipitate | Precipitate |
| Br− | 5303 | 273 | 149 |
| I− | >104, 860c | 455 | 95 |
| SO42− | 293d | Precipitate | Precipitate |
In an effort to compare the halide anion binding properties of the kinetically inert positively charged catenane 1·(PF6)2 receptor with a labile ion-pair receptor analogue, the heteroditopic XB bis-dibenzo-24-crown-8 functionalised 3,5-bis-iodopyridyl receptor XB11 was prepared for dibenzyl ammonium (DBA) cation-halide anion ion-pair binding investigation (see ESI† for synthetic details). Unfortunately, the poor solubility of XB11 in common organic solvents, including acetone and acetone/D2O mixtures, necessitated the 1H-NMR titration experiments being undertaken in 1:1 CD3CN/CDCl3. Monitoring perturbations of the receptor's aryl proton Ha in the vicinity of the XB recognition site, Bindfit analysis of the titration data (see ESI,† Table S1) determined the free XB11 receptor binds Br− (Ka = 3640 M−1) much more strongly than I− (Ka = 853 M−1). In the presence of two equivalents of DBA·PF6 resulting in pseudorotaxane assembly in each of the receptor's dibenzo-24-crown-8 substituents, TBA halide addition caused significant proton shifts, in particular in the aryl proton Ha of the XB recognition site and also induced chemical shift changes of proton signals of aryl protons of DBA+. Monitoring the aryl proton Ha proton, Bindfit analysis of the titration data (see ESI,† Table S1) determined 1:1 stoichiometric host–guest apparent association constant values for Br− and I− of 779 M−1 and 1752 M−1, respectively, indicating that co-bound secondary ammonium crown ether pseudorotaxane complexation enhances I− binding capability, and by stark contrast attenuates the strength of Br− binding. Tentatively, this may be a consequence of competitive DBA+ cation-bromide ion-pairing, which appears to be more significant than DBA+ cation-iodide ion-pair association in the mixed organic solvent medium.‡
In conclusion, a novel XB 3,5-bis-iodotriazole-pyridine receptor functionalised with secondary ammonium-dibenzo[24]crown-8 [2]catenane motifs (1·(PF6)2), was prepared in high yield via a post-synthetic MIM procedure. The kinetically inert nature of the mechanically interlocked ammonium cations facilitated XB-anion recognition in competitive aqueous-organic solvent mixtures of up to 20% D2O/d6-acetone with iodide. Importantly, 1·(PF6)2 displayed superior strong and selective anion binding affinity in comparison to labile sodium cation complexed bis-benzo[15]crown-5 XB and HB triazole-pyridine heteroditopic receptor analogues. These observations highlight the potential of exploiting secondary ammonium cation MIM crown ether encapsulation for augmenting anion recognition in competitive aqueous media.
T. B. thanks the Development and Promotion of Science and Technology Talents (DPST) Project, Thailand, for a full student scholarship. Y. C. T. thanks the Croucher Foundation for a scholarship. Authors thank Prof. Chutima Kuhakarn and Natthapat Sawekteeratana from Department of Chemistry, Mahidol University for spectroscopic characterisation of XB11.
Conflicts of interest
There are no conflicts to declare.References
- Q. He, G. I. Vargas-Zúñiga, S. H. Kim, S. K. Kim and J. L. Sessler, Chem. Rev., 2019, 119, 9753–9835 CrossRef CAS PubMed
.
- S. K. Kim and J. L. Sessler, Chem. Soc. Rev., 2010, 39, 3784–3809 RSC
- A. J. McConnell, A. Docker and P. D. Beer, ChemPlusChem, 2020, 85, 1824–1841 CrossRef CAS PubMed
- J. M. Mahoney, A. M. Beatty and B. D. Smith, Inorg. Chem., 2004, 43, 7617–7621 CrossRef CAS PubMed
- Q. He, G. M. Peters, V. M. Lynch and J. L. Sessler, Angew. Chem., Int. Ed., 2017, 56, 13396–13400 CrossRef CAS PubMed
- M. Li, B. Hua and F. Huang, Org. Chem. Front., 2021, 8, 3675–3680 RSC
- A. J. McConnell and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 5052–5061 CrossRef CAS PubMed
- B. Qiao, A. Sengupta, Y. Liu, K. P. McDonald, M. Pink, J. R. Anderson, K. Raghavachari and A. H. Flood, J. Am. Chem. Soc., 2015, 137, 9746–9757 CrossRef CAS PubMed
- X.-L. Ni, J. Tahara, S. Rahman, X. Zeng, D. L. Hughes, C. Redshaw and T. Yamato, Chem. – Asian J., 2012, 7, 519–527 CrossRef CAS PubMed
- M. Zakrzewski, D. Załubiniak and P. Piątek, Dalton Trans., 2018, 47, 323–330 RSC
- D.-H. Li and B. D. Smith, J. Org. Chem., 2019, 84, 2808–2816 CrossRef CAS PubMed
- J. Y. C. Lim, T. Bunchuay and P. D. Beer, Chem. – Eur. J., 2017, 23, 4700–4707 CrossRef CAS PubMed
- T. Bunchuay, A. Docker, A. J. Martinez-Martinez and P. D. Beer, Angew. Chem., Int. Ed., 2019, 58, 13823–13827 CrossRef CAS PubMed
- T. Bunchuay, K. Boonpalit, A. Docker, A. Ruengsuk, J. Tantirungrotechai, M. Sukwattanasinitt, P. Surawatanawong and P. D. Beer, Chem. Commun., 2021, 57, 11976–11979 RSC
- T. Bunchuay, A. Docker, N. G. White and P. D. Beer, Polyhedron, 2021, 209, 115482 CrossRef CAS
- T. Bunchuay, A. Docker, U. Eiamprasert, P. Surawatanawong, A. Brown and P. D. Beer, Angew. Chem., Int. Ed., 2020, 59, 12007–12012 CrossRef CAS PubMed
- P. R. Ashton, P. J. Campbell, P. T. Glink, D. Philp, N. Spencer, J. F. Stoddart, E. J. T. Chrystal, S. Menzer, D. J. Williams and P. A. Tasker, Angew. Chem., Int. Ed. Engl., 1995, 34, 1865–1869 CrossRef CAS
- S.-J. Rao, Q. Zhang, J. Mei, X.-H. Ye, C. Gao, Q.-C. Wang, D.-H. Qu and H. Tian, Chem. Sci., 2017, 8, 6777–6783 RSC
- H. V. Schröder, S. Sobottka, M. Nößler, H. Hupatz, M. Gaedke, B. Sarkar and C. A. Schalley, Chem. Sci., 2017, 8, 6300–6306 RSC
- G. Gholami, K. Zhu, G. Baggi, E. Schott, X. Zarate and S. J. Loeb, Chem. Sci., 2017, 8, 7718–7723 RSC
- H. Iwamoto, S. Tafuku, Y. Sato, W. Takizawa, W. Katagiri, E. Tayama, E. Hasegawa, Y. Fukazawa and T. Haino, Chem. Commun., 2016, 52, 319–322 RSC
- H. Iwamoto, W. Takizawa, K. Itoh, T. Hagiwara, E. Tayama, E. Hasegawa and T. Haino, J. Org. Chem., 2013, 78, 5205–5217 CrossRef CAS PubMed
- P. R. Aston, P. T. Glink, J. F. Stoddart, P. A. Tasker, A. J. P. White and D. J. Williams, Chem. – Eur. J., 1996, 2, 729–736 CrossRef
- H. R. Wessels and H. W. Gibson, Tetrahedron, 2016, 72, 396–399 CrossRef CAS
- S. T. Nguyen, L. K. Johnson, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1992, 114, 3974–3975 CrossRef CAS
- T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS PubMed
- S. W. Robinson and P. D. Beer, Org. Biomol. Chem., 2017, 15, 153–159 RSC
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC
- “BindFit v0.5|Supramolecular,” can be found under https://app.supramolecular.org/bindfit/.
- M. J. Power, D. T. J. Morris, I. J. Vitorica-Yrezabal and D. A. Leigh, J. Am. Chem. Soc., 2023, 145, 8593–8599 CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc03269h |
| ‡ Quantitative binding data could not be determined from Bindfit analysis of the TBACl titration isotherms. |
| This journal is © The Royal Society of Chemistry 2023 |