
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphorus derivatives of mesoionic carbenes: synthesis and characterization of triazaphosphole-5-ylidene → BF3 adducts†‡
Lea
Dettling
 a,
Niklas
Limberg
a,
Raphaela
Küppers
a,
Daniel
Frost
a,
Manuela
Weber
a,
Nathan T.
Coles
b,
Diego M.
Andrada
c and
Christian
Müller
*a
a,
Niklas
Limberg
a,
Raphaela
Küppers
a,
Daniel
Frost
a,
Manuela
Weber
a,
Nathan T.
Coles
b,
Diego M.
Andrada
c and
Christian
Müller
*a
aFreie Universität Berlin, Institute of Chemistry and Biochemistry, Fabeckstr. 34/36, Berlin 14195, Germany. E-mail: c.mueller@fu-berlin.de
bSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UK
cUniversität des Saarlandes, Anorganische Chemie, Saarbrücken 66123, Germany
First published on 26th July 2023
Abstract
Trimethylsilyl-substituted triazaphospholes were synthesized by a [3+2] cycloaddition reaction between organic azides and (CH3)3Si–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P. In an attempt to isolate their N-alkylated products, the formation of BF3 adducts of unprecedented triazaphosphol-5-ylidenes was found. The nature of the carboncarbene–boron bond was investigated within the DFT framework, revealing a strong donation of electrons from the carbene carbon atom to the boron atom combined with weak back-bonding.
P. In an attempt to isolate their N-alkylated products, the formation of BF3 adducts of unprecedented triazaphosphol-5-ylidenes was found. The nature of the carboncarbene–boron bond was investigated within the DFT framework, revealing a strong donation of electrons from the carbene carbon atom to the boron atom combined with weak back-bonding.
According to the isolobal relationship between a trivalent phosphorus atom and a C–H fragment, 3H-1,2,3,4-triazaphosphole derivatives A are phosphorus analogues of the well-known 1,4-disubstituted 1,2,3-triazoles B (Fig. 1), which play a prominent role in the field of “click”-chemistry.1 These heterocycles possess a high degree of aromaticity and can be obtained in a modular [3+2] cycloaddition reaction, starting from organic azides and phosphaalkynes.2,3
 | ||
| Fig. 1 Selected phosphorus and nitrogen based heterocycles and brief summary of this work (R/R′: alkyl-, aryl-group). | ||
Although triazaphospholes were first synthesized as early as 1984, reports of their coordination chemistry were first published almost 30 years later.3,4 Our group reported the coordination chemistry and photoluminescence properties of conjugated, pyridyl-functionalized triazaphospholes, bearing either tBu or Si(CH3)3-substituents at the 5-position of the heterocycle.5
Little is known about the chemical reactivity of triazaphospholes. In addition to our observation that they undergo cycloaddition–cycloreversion reactions with CF3CCCF3 forming C, we found that triazaphospholenium salts D are accessible by alkylation of triazaphospholes with Meerwein salts.6,7 Because a negatively charged carbon atom is valence isoelectronic to a phosphorus atom, these cationic phosphorus heterocycles are formally phosphorus congeners of the well-known mesoionic 1,2,3-triazolylidenes E and show an interesting coordination chemistry.7,8
Inspired by our recent investigations on 6-membered, 2-Si(CH3)3-substituted aromatic phosphorus heterocycles (phosphinines), we became interested in reinvestigating Si(CH3)3-substituted triazaphospholes (A, R′ = Si(CH3)3), particularly with respect to the formation of the corresponding triazaphospholenium salts (D, R′ = Si(CH3)3).5,9 As Si(CH3)3-groups linked to an aromatic system generally provide interesting electronic effects to the aromatic ring, we anticipated that these compounds might also undergo chemical transformations, such as protodesilylations or C–Si bond cleavage reactions.10
Much to our surprise, we now found that BF4−-salts of Si(CH3)3-substituted triazaphospholenium cations (G) undergo elimination of FSi(CH3)3 to form selectively BF3 adducts of unprecedented triazaphosphol-5-ylidenes (H). Interestingly, these heterocycles are the phosphorus congeners of tetrazol-5-ylidene carbenes (F) with an abnormal substitution pattern.11
The Si(CH3)3-substituted triazaphospholes 1a/b were synthesized by [3+2] cycloaddition reactions from Si(CH3)3–CP and benzyl azide (Bz-N3) or diisopropylphenyl azide (Dipp-N3), respectively in good yields (1a: 87%, 1b: 83%, Scheme 1). As expected, both compounds show resonances in the downfield region of the 31P{1H} NMR spectrum, at δ(ppm) = 214.8 (1a) and δ(ppm) = 222.9 (1b).
 | ||
| Scheme 1 Synthesis of the triazaphospholenium salts 2a/2b. | ||
Subsequently, we intended to convert 1a/b into the corresponding Si(CH3)3-substituted triazaphospholenium salts of type G (Fig. 1). Using an analogous methodology to that of the tBu-substituted derivatives, an equimolar mixture of compound 1a and [Me3O][BF4] was vigorously stirred in dichloromethane at T = 55 °C and the course of the reaction was followed by means of 31P{1H} NMR spectroscopy (Fig. S1, ESI†). After one hour, the formation of a new species was observed, which we attribute to the desired alkylated triazaphospholenium salt 2a, due to its characteristic chemical shift at δ(ppm) = 239.3.
However, considerable amounts of starting material were still present, while longer reaction times led to the formation of two more side-products. With the aim of preventing the formation of any side-products, [Me3O][BF4] was used in a slight excess and the reaction was kept at room temperature. After one day, 2a was obtained as the sole product in an isolated yield of 79% (Scheme 1). Similarly, the triazaphospholenium salt 2b was obtained in 97% isolated yield after washing the product with dry diethyl ether. 2b shows a resonance at δ(ppm) = 249.1 in the 31P{1H} NMR spectrum. In the 1H NMR spectrum of the triazaphospholenium salts, the introduced CH3-group can be detected as a characteristic singlet at δ(ppm) = 4.52 (2a) and δ(ppm) = 4.69 (2b), respectively.
Even though the Si(CH3)3-substituted triazaphospholenium salts 2a/b were finally synthesized in high yields, we were still wondering about the formation of the observed side products (vide supra), particularly at higher reaction temperatures. Consequently, we investigated the thermal stability of 2a by heating a solution of this compound in dimethoxyethane (DME) to T = 60 °C. Interestingly, the complete conversion of 2a to two new species (3a, 4a) was observed by 31P{1H} spectroscopy after one day.
We were able to separate both compounds by means of either extraction (3a) or flash column chromatography (4a). Compound 3a shows a resonance at δ(ppm) = 206.7 in the 31P{1H} NMR spectrum, which corresponds to a chemical shift difference of Δδ = 32.6 ppm compared to the starting material 2a (δ(ppm) = 239.3). Interestingly, a new signal can be observed in the 1H NMR spectrum at δ(ppm) = 9.26. This resonance appears as a doublet with a coupling constant of 2JH–P = 37.5 Hz, which is typical of protons in the α-position to a phosphorus atom. We therefore concluded that 3a must have been formed by protodesilylation of 2a, according to Scheme 2. The crystallographic characterization of 3a indeed confirmed that protodesilylation of 2a had occurred (Table S1, ESI†). Note, that the access to this compound would otherwise only be possible by cycloaddition reaction between an azide and hydrogen cyaphide (H–CP), or the cyaphido ligand (CP−), followed by a subsequent quaternization with Meerwein salt.12
 | ||
| Scheme 2 Formation of 3a and 4a upon heating of 2a in DME. | ||
Up to this point, the source of the proton still remains unknown. It should be noted, however, that protonated side-products have also been observed in the thermal degradation of free 1,2,3-triazol-5-ylidenes.13
Next, we turned our attention to the identification of the second species 4a. Surprisingly, this compound shows a quartet in the 19F{31P} NMR spectrum at δ(ppm) = −140.5 (q, 1JF–B = 37.7 Hz, Fig. S2, ESI†). Likewise, a quartet of doublets is observed in the corresponding 11B NMR spectrum at δ(ppm) = 0.6 (d, 2JB–P = 15.9 Hz, q, 1JB–F = 37.9 Hz). The chemical shifts and the coupling pattern is in line with the presence of a BF3-carbene adduct. For instance, the classical Arduengo carbene adduct IMes → BF3 (IMes = 1,3-dimesitylimidazol-2-ylidene) shows a resonance in the 19F NMR spectrum at δ(ppm) = −142.44 (q, 1JF–B = 34.6 Hz) and at δ(ppm) = −1.36 (q, 1JB–F = 34.6 Hz) in the corresponding 11B NMR spectrum.15 A coupling to the phosphorus nucleus in 4a would provide an additional splitting of the otherwise similar signals as can indeed be noticed in the 19F NMR and the 11B NMR spectra of 4a (19F NMR: δ(ppm) = −140.5 (d, 2JF–P = 14.6 Hz), 11B NMR: δ(ppm) = 0.6 ppm (d, 1JB–P = 15.9 Hz), Fig. S2, ESI†). Accordingly, the decoupled 11B{19F} NMR spectrum of 4a shows only a doublet due to the 1JB–P coupling (11B{19F} NMR: δ(ppm) = 0.7 (1JB–P = 15.8 Hz), Fig. S2, ESI†). Compound 4b was synthesized in an analogous manner.
Single crystals of 4a, suitable for X-ray diffraction, were obtained by layering a concentrated dichloromethane solution of 4a with n-pentane and the molecular structure of 4a, along with selected bond lengths and distances, is depicted in Fig. 2 (4b: Table S3, ESI†). The crystallographic characterization of 4a indeed reveals the presence of an abnormal carbene → BF3-adduct (Scheme 2). Compared to the protodesilylated triazaphospholenium salt 3a, the P(1)–C(1) bond in 4a is slightly elongated (4a: 1.711(2) Å; 3a: 1.706(2) Å), while the N(1)–P(1)–C(1) bond angle is somewhat larger and closer to 90° (4a: 87.31(8)°; 3a: 85.75(6)°). The C(1)–B(1) bond length of 1.640(3) Å) is very similar to the one found for the C–B-bond distance in the Lewis pairs of classical Arduengo carbenes (1.635(5) Å for IMes → BF3 and 1.669(6) Å for 4,5-dichloro-IMes → BF3).14 Similar bond lengths and distances were observed also for 4b (Table S3, ESI†). 4a can be described as a BF3 adduct of an unprecedented triazaphosphol-5-ylidene and thus as a phosphorus congener of the known tetrazol-5-ylidenes with an abnormal substitution pattern (F, Scheme 1).11 Moreover, 4a/b is again isoelectronic to the cationic part of the triazaphospholenium salt D (Fig. 1).
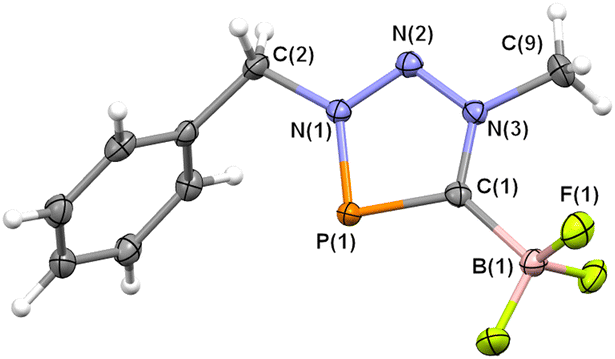 | ||
| Fig. 2 Molecular structure of 4a in the crystal. Displacement ellipsoids are shown at the 50% probability level. Selected experimental and theoretical [B3LYP-D3(BJ)/def2-SVP] bond lengths (Å) and angles (°): P(1)–C(1): 1.711(2) [1.722], P(1)–N(1): 1.705(2) [1.755], N(1)–N(2): 1.309(2) [1.301], N(2)–N(3): 1.320(2) [1.316], N(3)–C(1): 1.354(2) [1.357], C(1)–B(1): 1.640(3) [1.667], N(1)–P(1)–C(1): 87.31(8) [86.2]. | ||
From a mechanistic point of view, 4a/b is formed by elimination of FSiMe3 from 2a/b, which initially forms the carbene intermediate, that subsequently reacts with the remaining BF3. This would be in line with observations reported by Borozov and co-workers for 1-ethyl-3-methyl-1H-imidazolium BF4− (and respectively PF6−), which results in the formation of the corresponding NHC-based BF3 and PF5 Lewis pairs under rather harsh conditions.15 In this respect, 4a/b are also related to the GaCl3 adducts of neutral tetrazaphospholes, reported by Schulz and co-workers.16
In case of the formation of 4a/b the free carbene could not be observed spectroscopically, probably due to a rapid Lewis pair formation. However, the by-product FSiMe3 was detected by means of 1H–, 19F– and 29Si–1H-HMQC NMR spectroscopy and can easily be removed under vacuum. It is likely that the protonation of the carbene leads to the formation of 3a. This would be in agreement with observation that 1,2,3-triazlylidenes can undergo migration of an alkyl group, that is bound to the most nucleophilic nitrogen atom, to the carbene carbon atom, while the formation of a protodesilylated side-product is also observed.13
Because 3a and 4a are apparently formed by competing reactions, the isolated yields were moderate. We therefore anticipated targeted syntheses of both compounds. While other electrophiles, such as CH3I or CH3OTf, are not suitable for quaternization reactions at triazaphospholes, we chose triethyloxonium hexafluorophosphate instead. This indeed yielded the corresponding triazaphospholenium salts 5a/b in good isolated yields (5a: 80%, 5b: 93%, Scheme 3a). Compounds 5a/b provide again characteristic signals in the 31P{1H} NMR spectra at δ(ppm) = 238.1 (5a) and δ(ppm) = 249.0 (5b), respectively. Additionally, the introduced ethyl group shows typical signals in the 1H NMR spectra (5a: δ(ppm) = 4.73 (q, J = 7.5 Hz), 1.74 (t, J = 7.3 Hz,); 5b: δ(ppm) = 4.84 (q, J = 7.1 Hz), 1.69 (t, J = 7.1 Hz)).
 | ||
| Scheme 3 Synthesis of the triazaphospholenium salts 5a/5b (a). Preparation of the BEt3-adduct 7b and the protodesilylated compound 6b (b). | ||
We also anticipated that the addition of potassium fluoride (KF) in the presence of a Boron-based Lewis acid BR3 should facilitate the formation of FSiMe3 and the abnormal carbene → BR3 adduct, while K[PF6] might precipitate from the solution. For this purpose, BH3·SMe2, BH3·THF, BF3·SMe2, BF3·OEt2, and BEt3, were used in combination with 5b. Interestingly, except for BEt3, the selective formation of the protodesilylated product 6b was observed, most likely due to contaminations with H2O/HF. On the other hand, in the presence of BEt3, the BEt3-adduct 7b was selectively formed (Scheme 3b). This was confirmed by an X-ray structural analysis (Fig. 3). Compound 6b was also independently synthesized and characterized crystallographically (see Table S4, ESI†).
 | ||
| Fig. 3 Molecular structure of 7b in the crystal. Displacement ellipsoids are shown at the 50% probability level. Only one independent molecule in the asymmetric unit is shown. Selected bond lengths (Å) and angles (°): P(1)–C(1): 1.7306(10), P(1)–N(1): 1.7110(9), N(1)–N(2): 1.3146(12), N(2)–N(3): 1.3310(12), N(3)–C(1): 1.3629(13) C(1)–B(1): 1.6468(15); N(1)–P(1)–C(1): 88.26(4). | ||
Finally, we investigated the nature of the Ccarb → B bond by means of DFT calculations at the B3LYP-D3(BJ)/def2-SVP level of theory (see ESI† for details). We were particularly interested in the effect of the phosphorus atom on the donor–acceptor abilities of the abnormal carbene moiety.17 The optimized structures are in good agreement with the experimentally measured ones (Fig. 2, 3 and Table S3 and Fig. S43, ESI†), with the Ccarb–B bond lengths being slightly longer than those observed experimentally (4a 1.667 Å and 4b 1.668 Å). The natural bond orbital analysis (Table S6 and Fig. S44, ESI†) indicates a strong donation of electrons as the BF3 bears a negative partial charge of −0.47 (4a) and −0.46 (4b). The Wiberg bond orders indicate a single bond character with 0.71 au in 4a and 4b. The Bader topological analysis18 reveals an electron accumulation between the carbene carbon atom and the boron atom with a bond critical point that possesses a relatively small electron density value (ρ(r) 0.97 e Å−34a, 0.96 e Å−34b, and 0.92 e Å−37b) with negative Laplacian values of ∇2ρ(r) = −1.82 e Å−5 (4a) and ∇2ρ(r) = −1.77 e Å−5 (4b), respectively a positive value of ∇2ρ(r) = 3.74 e Å−5 for 7b (Fig. 4). Additionally, we have performed energy decomposition analysis (EDA-NOCV)19 to quantitatively assess the chemical bonding situation in these adducts, taking as fragments the neutral triazaphosphol-5-ylidenes (4a/b, 7b) and BF3 moieties (Table S7 and Fig. S45, ESI†). The bond dissociation energy is slightly weaker than in other borane adducts, with energies of 38.4 (4a), 37.5 (4b) and 40.0 (7b) kcal mol−1.20 However, the dissection into preparation energy and interaction energy suggests that the geometrical deformation of BF3 or BEt3 brings a significant energy penalty (31.0 and 26.0 kcal mol−1).
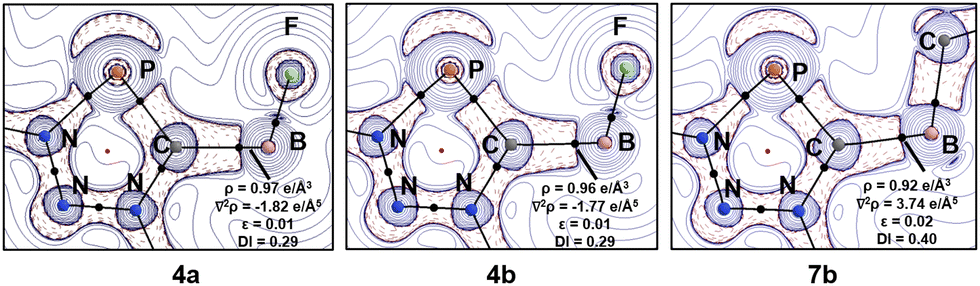 | ||
| Fig. 4 Laplacian distribution of the electron density of compounds 4a/b and 7b (B3LYP-D3(BJ)/def2-TZVP//B3LYP-D3(BJ)/def2-SVP). Contour line diagrams of the Laplacian distribution ∇2ρ(r) in the PHC ring plane. Dashed red lines indicate areas of charge concentration (∇2ρ(r) < 0) while solid blue lines show areas of charge depletion (∇2ρ(r) > 0). The thick solid lines connecting the atomic nuclei are the bond paths and the small dots are the critical points. Bond Critical Points (in black), Ring Critical Points (in red). | ||
The interaction energy is comparable to other known Ccarbene → B adducts, counting −72.1 (4a), −70.7 (4b) and −68.4 (7b) kcal mol−1.21 Further decomposition reveals an ionic nature of the bond with approximately 53% electrostatic interaction and 45% orbital interaction. Note that the electrostatic interaction refers to the electrostatic attraction between the charge distribution of the fragments, differentiating from the VB ionic bonds (for further details see ref. 20). The orbital term can be analysed with the Natural Orbitals for Chemical Valence (NOCV), where the σ-donation counts for ∼80% of the orbital interaction, while the π-back donation is only ∼5%. Fig. S46 (ESI†) shows a comparison of the frontier molecular orbital energies of the model systems 1,2,3-triazolylidene, tetrazol-5-ylidene and triazaphosphole-5-ylidene.
The energy of the σ-lone pair on the carbene carbon atom is comparable to the triazole analogue and agrees with the strong donor properties towards BF3 or BEt3. On the other hand, the presence of a phosphorus atom provides less stabilization of the π* orbital, leading to relatively high π-acceptor properties.
In summary, we have found an access to BF3 adducts of hitherto unknown triazaphosphol-5-ylidenes, which are the phosphorus analogs of tetrazol-5-ylidene carbenes with abnormal substitution pattern. Quantum chemical calculations reveal a strong σ-donor property and marginal π-accepting ligand properties. The access to transition metal complexes containing our novel triazaphosphole-5-ylidenes as ligands are currently performed in our laboratories.
Financial support provided by Freie Universität Berlin and the Deutsche Forschungsgemeinschaft (DFG) are gratefully acknowledged. D. M. Andrada thanks Prof. Dr. David Scheschkewitz and the University of Saarland for generous support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Phosphorus: The Carbon Copy: From Organophosphorus to Phospha-Organic Chemistry, ed. K. B. Dillon, F. Mathey and J. F. Nixon, John Wiley & Sons, 1998 Search PubMed; (b) J. A. W. Sklorz and C. Müller, Eur. J. Inorg. Chem., 2016, 595 CrossRef CAS.
- L. Nyulászi, T. Veszprémi, J. Réffy, B. Burkhardt and M. Regitz, J. Am. Chem. Soc., 1992, 114, 9080 CrossRef.
- (a) Y. Y. C. Yeung Lam, Ko, R. Carrié, A. Muench and G. Becker, J. Chem. Soc., Chem. Commun., 1984, 1634 Search PubMed; (b) W. Rösch and M. Regitz, Angew. Chem., Int. Ed. Engl., 1984, 23, 900 CrossRef.
- (a) S. L. Choong, A. Nafady, A. Stasch, A. M. Bond and C. Jones, Dalton Trans., 2013, 42, 7775 RSC; (b) S. L. Choong, C. Jones and A. Stasch, Dalton Trans., 2010, 39, 5774 RSC.
- J. A. W. Sklorz, S. Hoof, N. Rades, N. De Rycke, L. Könczöl, D. Szieberth, M. Weber, J. Wiecko, L. Nyulászi, M. Hissler and C. Müller, Chem. – Eur. J., 2015, 21, 11096 CrossRef CAS PubMed.
- L. Dettling, M. Papke, J. A. W. Sklorz, D. Buzsáki, Z. Kelemen, M. Weber, L. Nyulászi and C. Müller, Chem. Commun., 2022, 58, 7745 RSC.
- M. Papke, L. Dettling, J. A. W. Sklorz, D. Szieberth, L. Nyulászi and C. Müller, Angew. Chem., Int. Ed., 2017, 56, 16484 CrossRef CAS PubMed.
- G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 4759 CrossRef CAS PubMed.
- M. H. Habicht, F. Wossidlo, T. Bens, E. A. Pidko and C. Müller, Chem. – Eur. J., 2018, 24, 944 CrossRef CAS PubMed.
- M. Blug, O. Piechaczyk, M. Fustier, N. Mézailles and P. Le Floch, J. Org. Chem., 2008, 73, 3258 CrossRef CAS PubMed; Y. Hatanaka and T. Hiyama, J. Org. Chem., 1988, 53, 918 CrossRef.
- (a) W. P. Norris and R. A. Henry, Tetrahedron Lett., 1965, 17, 1213 CrossRef; (b) J. Müller, K. Öfele and G. Krebs, J. Organometal. Chem., 1974, 82, 383 CrossRef; (c) L. Schaper, X. Wei, P. J. Altmann, K. Öfele, A. Pöthig, M. Drees, J. Mink, E. Herdtweck, B. Bechlars, W. A. Herrmann and F. E. Kühn, Inorg. Chem., 2013, 52, 7031 CrossRef CAS PubMed.
- (a) W. J. Transue, A. Velian, M. Nava, M. A. Martin-Drumel, C. C. Womack, J. Jiang, G. L. Hou, X. Bin Wang, M. C. McCarthy, R. W. Field and C. C. Cummins, J. Am. Chem. Soc., 2016, 138, 6731 CrossRef CAS PubMed; (b) T. E. Gier, J. Am. Chem. Soc., 1961, 83, 1769 CrossRef CAS; (c) E. S. Yang, A. Mapp, A. Taylor, P. D. Beer and J. Goicoechea, Chem. – Eur. J., 2023, e202301648 CrossRef PubMed; T. Görlich, D. Frost, N. Boback, N. T. Coles, B. Dittrich, P. Müller, W. D. Jones and C. Müller, J. Am. Chem. Soc., 2021, 143, 19365 Search PubMed.
- (a) G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 4759 CrossRef CAS PubMed; (b) J. Bouffard, B. K. Keitz, R. Tonner, G. Guisado-Barrios, G. Frenking, R. H. Grubbs and G. Bertrand, Organometallics, 2011, 30, 2617 CrossRef CAS PubMed.
- A. J. Arduengo III, F. Davidson, R. Krafczyk, W. J. Marshall and R. Schmutzler, Monatsh. Chem., 2000, 131, 251 CrossRef.
- C. Tian, W. Nie, M. V. Borzov and P. Su, Organometallics, 2012, 31, 1751 CrossRef CAS.
- A. Villinger, P. Mayer and A. Schulz, Chem. Commun., 2006, 1236 RSC.
- D. Munz, Organometallics, 2018, 37, 275 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Clarendon, Oxford, 1990 Search PubMed.
- L. Zhao, M. von Hopffgarten, D. M. Andrada and G. Frenking, WIREs Comput. Mol. Sci., 2018, 8, e13450 Search PubMed.
- S. Dutta, S. M. De, S. Bose, E. Mahal and D. Koley, Eur. J. Inorg. Chem., 2020, 638 CrossRef CAS.
- (a) A. Krapp, F. M. Bickelhaupt and G. Frenking, Chem. – Eur. J., 2006, 12, 9196 CrossRef CAS PubMed; (b) I. Fernández, N. Holzmann and G. Frenking, Chem. – Eur. J., 2020, 26, 14194 CrossRef PubMed; (c) L. Zhao, S. Pan and G. Frenking, J. Chem. Phys., 2022, 157, 034105 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Peter Jutzi on the occasion of his 85th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2279453–2279457. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc03268j |
| This journal is © The Royal Society of Chemistry 2023 |