
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual photoredox nickel-catalyzed silylation of aryl/heteroaryl bromides using hydrosilanes†
Shuai
Liu
,
Frédéric
Robert
 and
Yannick
Landais
*
and
Yannick
Landais
*
University Bordeaux, CNRS, Bordeaux INP, ISM, UMR 5255, F-33400 Talence, France. E-mail: yannick.landais@u-bordeaux.fr
First published on 28th August 2023
Abstract
Dual Ni and Ir catalysis enables the preparation of arylsilanes having a (TMS)3Si substituent from the corresponding aryl bromides and (TMS)3SiH at 30 °C using visible-light irradiation. This protocol avoids strong bases, high temperature and air and moisture sensitive silyl reagents, providing the expected arylsilanes in moderate to good yields. The reaction was shown to proceed through a silyl radical, likely generated by hydrogen atom abstraction from (TMS)3SiH by a bromide radical.
Arylsilanes have found widespread utility in organic synthesis and materials chemistry due to their unique reactivity and properties.1 They may also serve as versatile intermediates in organic transformations, including electrophilic ipso substitution,2 Heck,3 and Hiyama–Denmark cross-coupling reactions.4 These transformations enable the introduction of aryl groups onto diverse substrates, making arylsilanes valuable building blocks for the synthesis of complex organic molecules. Arylsilanes exhibit remarkable stability, the C–Si bond being highly resistant to oxidation and hydrolysis, allowing for synthetic manipulations under harsh conditions without compromising the integrity of the C–Si bond. In addition to their synthetic utility, their unique properties, such as thermal stability and low toxicity, make them suitable for use in polymers and electronic materials.5 Overall, arylsilanes offer a valuable toolbox for synthetic chemists, providing versatile reactivity, stability, and tunable properties. Their applications span across diverse fields, including synthesis, materials science, and pharmaceutical research,6 making them important components in modern organic chemistry.1,2
The traditional approach of building arylsilanes involves the metalation through a Barbier-type reaction or lithium–halogen exchange using an organo-lithium reagent followed by a silylation of the resulting aryl-Mg(Li) intermediate (Scheme 1a).1,7 This strategy however has several drawbacks, including low functional group tolerance, harsh reaction conditions, and requires the use of air- and moisture sensitive reagents. Transition-metal silylation of aryl halides has offered an attractive alternative to this strategy, allowing the Csp2–Si bond formation starting from a broad variety of aryl(heteroaryl) halides and various silylating agents (Scheme 1b).8 Amongst these reagents hydrosilanes and silylboranes, have found a widespread use although the former tends to provide various amount of reduced products, while the latter are air and moisture sensitive. Disilanes have also been used, but suffer from a low reactivity of the Si–Si bond, which activation requires strong bases and high temperature. Recently sodium silanolates and silylsilanolates were designed to address some of these issues.9
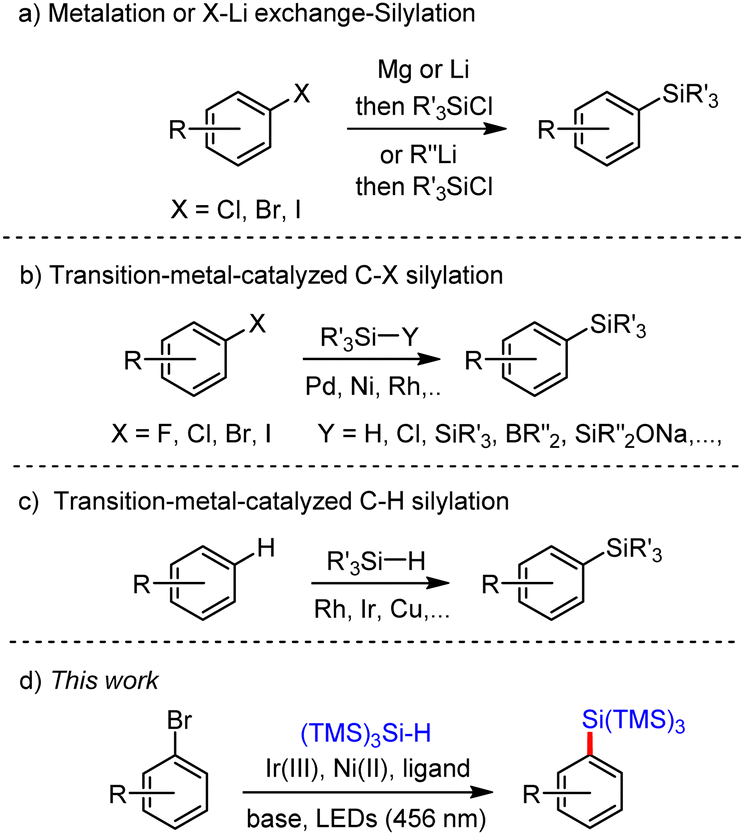 | ||
| Scheme 1 Cross-coupling reactions between aryl halides or arenes and silylating reagents. | ||
Friedel–Crafts C–H silylation is a powerful technique, which is however restricted to electron-rich arenes and heteroarenes such as indoles and pyrroles.10 Recently, transition metal-catalyzed cross-dehydrogenative coupling between arenes/heteroarenes and hydrosilanes to deliver arylsilanes has grown in popularity (Scheme 1c).11 Iridium, ruthenium and rhodium complexes were shown to exhibit efficient catalytic activities in C–H silylation.12 However, the following challenges nevertheless continue to exist: (1) low regioselectivity; (2) need for directing groups, noble metal catalysts, and hydrogen acceptors. More recently, Liu and co-workers disclosed a radical mediated site-selective aryl/heteroaryl C–H silylation in the presence of Cu2O catalyst, a peroxide, and a hydrosilane.13 This strategy exhibits exceptional para-selectivity. However, large excess of peroxide, hydrosilane, and high temperatures are required. Finally, unactivated arenes and heteroarenes were recently silylated using silyldiazenes under transition-metal free conditions.14 In this context, we recently uncovered a photoredox mediated dual Ir/Ni catalysis15 for the construction of arylsilanes, which proceeds at room temperature, using readily available tristrimethylsilylsilane ((TMS)3SiH)16 as the silicon source and aryl bromides as aryl precursors (Scheme 1d).
The process was optimized using methyl-4-bromobenzoate 1a as model substrate and TMS3SiH as the silicon source. Following extensive screening, we discovered that [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 as a photocatalyst and NiBr2 as a synergistic catalyst, sodium carbonate (Na2CO3) as the base at room temperature in acetone, delivered the product in reasonable yield (entry 1). Several solvents were also used to optimize the reaction conditions (SI), but “green” (dry) acetone proved to be the best. Two side-products were identified, including biaryl 3a and the debrominated adduct 4a, which could be easily discarded upon chromatography. Stronger inorganic bases, such as K2CO3 and K3PO4 were tested, but led to decomposition of the starting material and to very low yield in 2a (entries 2 and 3). The organic base 2,6-lutidine efficiently yielded the corresponding product, albeit in a slightly lower yield (entry 4). The nature of the Ni(II) catalyst was also studied. NiCl2·glyme, NiBr2·glyme, and NiBr2 all afforded comparable yields. NiBr2 being the cheapest available, it was retained for the remaining part of the study (entries 1, 5 and 6). Next, several ligands were also screened, with dtbbpy providing the best yields (entries 7 and 8). Interestingly, when 5,5′-CF3-bpy was employed as a ligand, the self-coupling product 3a was the main product with a 47% yield (entry 9). The amount of (TMS)3SiH in the reaction proved to be crucial as shown with the better yield obtained when 3 equivalents was used (entry 10). Under these optimized conditions, absence or traces of 2a were observed when the reaction was performed in the absence of a catalyst (entry 11), light (entry 12) or base (entry 13). The target product was then produced in 45% yield when the organic photocatalyst 4-CzIPN was utilized in place of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (entry 14). Other silanes including PhMe2SiH were tested, but led to much lower yields (<10%) under these conditions. Aryl chlorides were also used as precursors but were found to be poorly reactive.
Having established the optimal conditions (Table 1, entry 10), various bromides were then submitted to the Ir/Ni cross-coupling process. The corresponding arylsilanes were generated in moderate to good yields, as shown in Scheme 2. Benzene rings with electron-deficient substituents, including esters and ketones as in 2a, 2b and 2p–r, nitriles (2c, 2d), amides (2h, 2i, 2s), sulfones (2j), halogens (2f, 2g), and trifluoromethyl groups (2e), reacted smoothly. Interestingly, the method is compatible with the presence of acidic protons as in 2h, 2i or 2s. The formation of ortho-cyanoarylsilane 2d in only moderate yield demonstrates that steric hindrance around the reacting center may be a limiting factor. On the other hand, attempting the reaction with methyl 2-bromobenzoate as the substrate did not result in any reaction (ESI†). Certain electron-donating substituents, such as methyl (2n), ethyl (2m), and t-butyl (2o), are also compatible with this system, although the yield is lower, most likely due to the ease with which electron-rich aromatic rings are oxidized by the Ir photocatalyst in its excited-state. Benzylic positions as in 2i remains however intact. It is worth noting that derivatives of certain natural products (2p–s) exhibit efficient reactivity, yielding the desired product in moderate to good yields. To our satisfaction, although the yield was modest, we found that heteroaromatics (i.e.2t–v) were also appropriate substrates for this reaction conditions. Finally, we attempted to extend the methodology to alkyl bromides. Primary alkyl bromides were thus shown to react to afford the corresponding products (2w–y) in moderate yields.
|
|
||||
|---|---|---|---|---|
| Entry | Deviation from standard conditionsa | Yieldc (%) 2a | Yieldc (%) 3a | Yieldc (%) 4a |
| a Standard conditions: [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (2 mol%), NiBr2 (5 mol%), dtbbpy (10 mol%), Na2CO3 (2 eq.), 1a (0.2 mmol, 1 eq.), (TMS)3SiH (0.4 mmol, 2 eq.), dry acetone (2.0 mL), Kessil 40 W LEDs (456 nm), 30 °C, 20 h. b (TMS)3SiH (0.6 mmol, 3 eq.). c 1H NMR yields of 2a, 3a, and 4a using mesitylene as an internal standard. | ||||
| 1 | None | 44 | 5 | 12 |
| 2 | K2CO3 | <10 | — | — |
| 3 | K3PO4 | <10 | — | — |
| 4 | 2,6-Lutidine | 37 | 4 | 36 |
| 5 | NiCl2·glyme | 42 | 16 | 30 |
| 6 | NiBr2·glyme | 44 | 17 | 20 |
| 7 | bpy | 38 | 24 | 25 |
| 8 | 4,4′-OMe-bpy | 21 | 6 | 32 |
| 9 | 5,5′-CF3-bpy | 17 | 47 | 11 |
| 10 | (TMS)3SiH (3 eq.) | 62 | 11 | 14 |
| 11b | No catalyst [Ir] | 0 | 0 | 0 |
| 12b | No light | 0 | 0 | 0 |
| 13b | No base | Traces | Traces | Traces |
| 14b | 4-CzIPN instead of [Ir] | 45 | 7 | 11 |
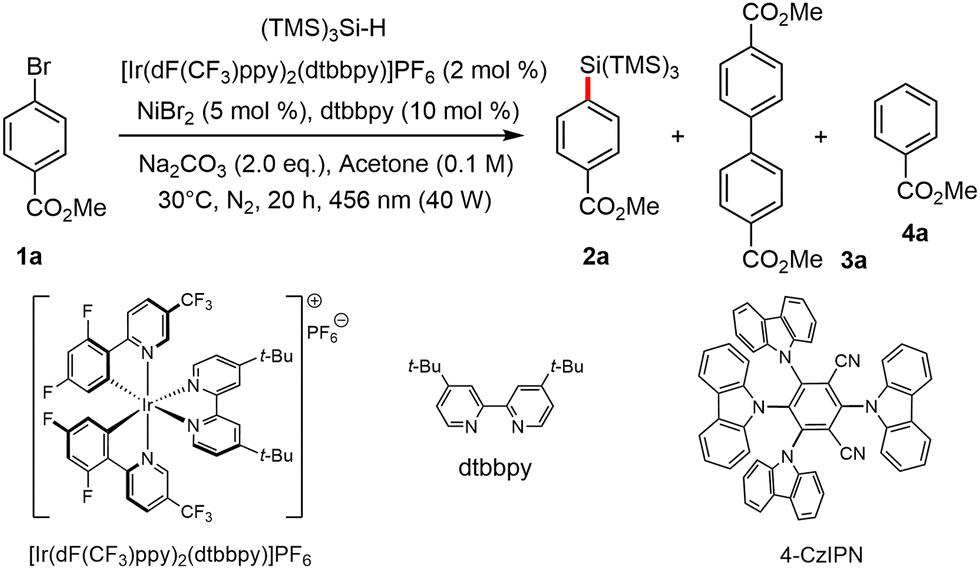
 | ||
| Scheme 2 Scope of the Ir/Ni-catalyzed cross-coupling process between bromoarenes 1 and (TMS)3SiH. | ||
Preliminary mechanistic investigations were also performed. For instance, when the reaction between 1a and (TMS)3SiH under the optimal conditions above was repeated in the presence of TEMPO, no reaction occurred, indicating the occurrence of radical species during the process (Scheme 3, eqn (1)). Benzyl acrylate was then added to the standard reaction mixture as a radical trap. Under these conditions, the target product 2a was not detected, but addition product 5 was isolated in good yield, diagnostic of the formation of a silyl radical, which is known to add efficiently onto electron-poor olefins (Scheme 3, eqn (2)).17 When the reaction was repeated in the presence of 1,1-diphenylethylene, the desired product 2a was not formed, but the hydrosilylation product 6 could be detected by GC-MS, confirming the presence of a silyl radical (Scheme 3, eqn (3)). Finally, when both aryl bromides 1a and 1m were mixed together under the above dual catalytic system, arylsilanes 2a and 2m, reduced products 4a and 4m but also homocoupling products 3a and 3m were detected by GC-MS. Heterobiaryl coupling product 7 was also found, suggesting the formation of aryl radicals during the process (Scheme 3, eqn (4)).18
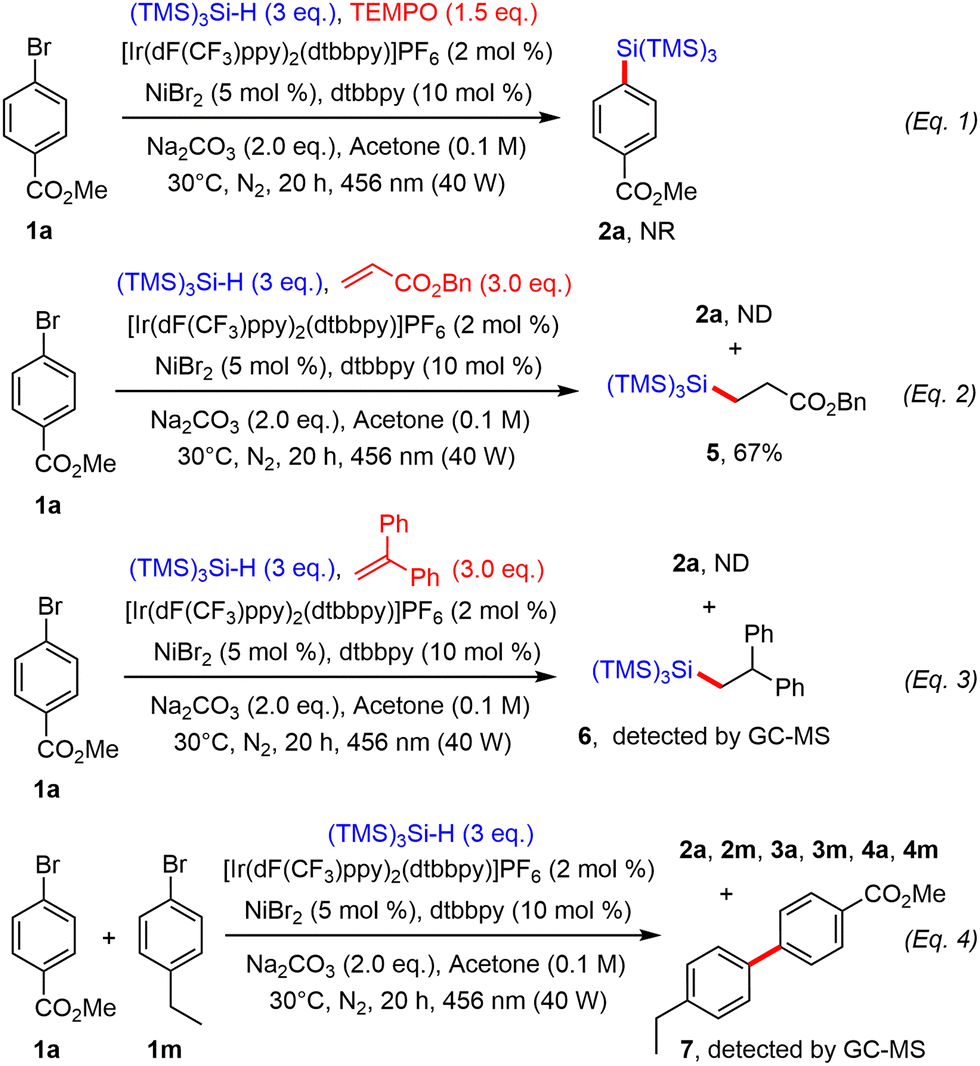 | ||
| Scheme 3 Mechanistic studies. | ||
Based on these experimental evidences and literature precedent using similar dual catalytic systems,15b a plausible mechanism was proposed (Fig. 1). The Br− issued from NiBr2 is oxidized by Ir(III)* in its excited-state, under blue-light irradiation, to give Br˙ and Ir(II).15b Oxidation of electron-rich aryl bromides by Ir(III)* may compete with this step, explaining the lower yield with these arenes (ESI†). Br˙ then abstracts a hydrogen from the silane to afford the silyl radical I.17 Meanwhile, the Ni(I) is reduced by the resulting Ir(II) into Ni(0) and the photoactivable Ir(III). Oxidative addition of aryl bromide 1 onto the Ni(0) complex provides the Ni(II) species II and Br−. The silyl radical I then reacts with II, leading to the Ni(III) complex III, the reductive elimination of which affording the final coupling product 2. Interestingly, the silyl radical might also abstract a bromine atom for aryl bromide 1 and afford an aryl radical IV,18 which could enter into the Ni catalytic cycle, reacting for instance with II to provide after reductive elimination a biaryl 3. Such a pathway would explain the formation of 3a (Table 1). The same aryl radical IV may react further with (TMS)3SiH to furnish reduction product 4.16,19 Abstraction of the bromide from 1 to generate unstable aryl radical IV, is likely a slower pathway as compared with the reaction of the (TMS)3Si radical I with intermediate II.19
 | ||
| Fig. 1 Dual catalytic system. Suggested mechanism. | ||
In summary, we report here on a new silylation process of arylbromides using simple (TMS)3SiH. The combination of the [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 complex and NiBr2/dtbpy catalyst was shown to trigger the formation of aryl/heteroaryl/alkyl silanes using commercially available (TMS)3SiH as a silicon source.16,17 This approach exhibits good functional group tolerance, regardless of their electronic properties, leading to the desired products with moderate to satisfying yields. In addition, not only aryl bromides and heteroaryl bromides, but also primary alkyl bromides can serve as suitable substrates. This strategy offers a complementary method to access these synthetically important synthons, avoiding strongly basic organo-lithiums and sensitive chlorosilanes. It also offers an entry for the late stage functionalization of pharmaceutically relevant compounds20 and access to silicon isosteres through the so-called “silicon-switch” strategy.21 Work in now under way to further extend this method to other aryl derivatives and silanes.
SL thanks the Chinese Scholarship Council (CSC) for a PhD grant. We are grateful to the University of Bordeaux (UBx) and to the CNRS for financial support. Cesamo (Ubx) is gratefully acknowledged for GC-MS experiments.
Conflicts of interest
There are no conflicts to declare.Notes and references
-
(a)
B. A. Key, Arylsilanes in Science of Synthesis in Houben-Weyl methods of molecular transformations, Compounds of group 15 (As, Sb, Bi) and silicon compounds, ed. I. Fleming, 2002, vol. 4, p. 685 Search PubMed
; (b) M. A. Brook, Silicon in Organic, Organometallic, and Polymer Chemistry, Wiley, New York, 2000 Search PubMed
-
(a) B. Bennetau and J. Dunogues, Synlett, 1993, 171 CrossRef CAS
- R. C. Larock, E. K. Yum and M. D. Refvik, J. Org. Chem., 1998, 63, 7652 CrossRef CAS
-
(a)
T. Hiyama in Metal-catalyzed Cross-Coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weinheim, 1998, p. 421 Search PubMed
- D. Sun, Z. Ren, M. R. Bryce and S. Yan, J. Mater. Chem. C, 2015, 3, 9496 RSC
- R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779 CrossRef CAS PubMed
-
E. W. Colvin, Silicon reagents in organic synthesis, Academic press, 1990, p. 39 and references therein Search PubMed
-
(a) For a review, see: Z. Xu, W.-S. Huang, J. Zhang and L.-W. Xu, Synthesis, 2015, 3645 CrossRef CAS
-
(a) H. Yamagishi, H. Saito, J. Shimokawa and H. Yorimitsu, ACS Catal., 2021, 11, 10095 CrossRef CAS
-
(a) U. Frick and G. Simchen, Synthesis, 1984, 929 CrossRef CAS
-
(a) C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946 CrossRef CAS PubMed
-
(a) B. Lu and J. R. Falck, Angew. Chem., Int. Ed., 2008, 47, 7508 CrossRef CAS PubMed
- Z. B. Xu, L. Chai and Z.-Q. Liu, Org. Lett., 2017, 19, 5573 CrossRef CAS PubMed
- B. Neil, F. Lucien, L. Fensterbank and C. Chauvier, ACS Catal., 2021, 11, 13085 CrossRef CAS
-
(a) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485 CrossRef CAS PubMed
- C. Chatgilialoglu, C. Ferreri, Y. Landais and V. I. Timokhin, Chem. Rev., 2018, 118, 6516 CrossRef CAS PubMed
-
C. Chatgilialoglu, Organosilanes in Radical Chemistry, Wiley, Chichester, UK, 2004 Search PubMed
- N. Kvasovs and V. Gevorgyan, Chem. Soc. Rev., 2021, 50, 2244 RSC
- J. J. Devery, J. D. Nguyen, C. Dai and C. R. J. Stephenson, ACS Catal., 2016, 6, 5962 CrossRef CAS
- R. Cannalire, S. Pelliccia, L. Sancineto, E. Novellino, G. C. Tron and M. Giustiniano, Chem. Soc. Rev., 2021, 50, 766 RSC
-
R. Tacke and S. Dörrich, Drug Design Based on the Carbon/Silicon Switch Strategy, in Atypical Elements in Drug Design, ed. J. Schwarz, Topics in Medicinal Chemistry, 2016, vol. 17, p. 29 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc03246a |
| This journal is © The Royal Society of Chemistry 2023 |