
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn in-cell spin-labelling methodology provides structural information on cytoplasmic proteins in bacteria†
Yulia
Shenberger‡
,
Lada
Gevorkyan-Airapetov‡
,
Melanie
Hirsch
,
Lukas
Hofmann
and
Sharon
Ruthstein
 *
*
Department of Chemistry, Faculty of Exact Sciences and Institute of Nanotechnology and Advanced Materials, Bar Ilan university 5290002, Israel. E-mail: Sharon.ruthstein@biu.ac.il
First published on 7th August 2023
Abstract
EPR in-cell spin-labeling was applied to CueR in E. coli. The methodology employed a Cu(II)-NTA complexed with dHis. High resolved in-cell distance distributions were obtained revealing minor differences between in vitro and in-cell data. This methodology allows study of structural changes of any protein in-cell, independent of size or cellular system.
Understanding structural dynamics of proteins within their physiological environment can be considered as the Holy Grail of structural biology. Currently, the most common methods used for resolving protein structures within the cell are nuclear magnetic resonance (NMR), Förster resonance energy transfer (FRET), and cryo electron tomography (cryo-ET)). These fast-advancing technologies provide different perspectives on spatial-temporal resolution and structural rearrangements of proteins within the cell. In cell NMR can detect interactions between proteins and small molecules and provides three dimensional structures about the proteins of interest,1,2 yet it is limited by the size of the biological system of interest. In cell FRET can report on dynamical changes of proteins in the cell3,4 but has not yet provided accurate structural information. Cryo-ET is an excellent tool for obtaining structural information on large symmetric biological systems and complexes.6 At the same time, it is less preferred for monitoring proteins of low abundance or low symmetry within a cell.7,8
During the last decade, in cell electron paramagnetic resonance (EPR) spectroscopy has emerged as an excellent methodology to follow biological mechanisms at high resolution within the cell.9–16 EPR distance measurements, such as double electron electron resonance (DEER), can define distances within a biological system in the nanometer range (1.5–10.0 nm).17 EPR spectroscopy offers numerous advantages over other biophysical tools. First, its high sensitivity, EPR can target biomolecules present at concentrations ranging from the micromolar level to the few tens nanomolar level.18 Moreover, EPR measurements are not limited by the size or complexity of the biological system nor by the environment in which it is found. However, EPR spectroscopy requires paramagnetic centers, a need that raises several challenges, especially for in cell EPR experiments. The first obstacle is that the selected spin-label should be stable in a reducing environment, such as the cytoplasm. Therefore, the most common spin-labels used for in cell EPR measurements are Gd(III)-19 and trityl-20–23 based spin-labels. A second obstacle is that the currently applied spin-labeling methodology requires delivery of the spin-labeled protein into the cell, after the spin-labeling procedure was performed outside of the cell. This limits the size of the biomolecule of interest, as well as the cellular system that can be investigated, which is limited by how much the cell membrane can be distorted. As such, this method is mostly employed for studies of eukaryotic systems.9,12,14,24
Previously, a genetically encoded nitroxide spin labeling methodology was suggested on overexpressed proteins in E. coli, showing a sufficient efficiency of overexpressed spin-labeled protein.16 Here we report the development of a new in cell spin-labeling methodology. The advantage of this method is that it is performed on over-expressed proteins within the bacterial cell, and with the spin-labeling process being carried out within the cell itself. Using this method, any protein, regardless of size and/or complexity, can be studied in a variety of cellular systems. This approach uses Cu(II)-nitriloacetic acid (Cu(II)-NTA) as the spin-label. Cu(II)-NTA shows high-affinity to dHis sites, especially for dHis sites located within helices (Fig. 1A).18,25–27 In the cell, free Cu(II) is immediately reduced to the diamagnetic state, Cu(I). The NTA ligand ensures that Cu(II) will not be reduced in the cytosol.27–30 To form a stable complex, the two histidine residues of such sites should be separated by four amino acids to ensure high Cu(II)-NTA binding.7,31 The two histidine residues are required to be in the plane or in a 360° turn to complex Cu(II) which is about four amino acids apart.32 Since this spin-labeling method is performed on the protein backbone, it provides very narrow distance distribution functions, and can differentiate between minor conformational changes of the protein. As proof of concept of this methodology, the copper-sensitive transcription factor CueR (Fig. 1B)33–40 was employed. CueR is a transcription factor that prevents copper toxicity in Gram-negative bacteria. It possesses high affinity to the reduced form of copper, Cu(I). Upon Cu(I) binding in a linear coordination to C112 and C120 residues, CueR initiates a transcription process that leads to the expression of proteins that either oxidize copper to the less toxic Cu(II) form or shuttle copper outside the cell. We previously showed that DEER measurements performed on a CueR_L60H_G64H mutant labeled with Cu(II)-NTA can follow conformational changes of the protein as a function of Cu(I) and DNA binding in vitro.34 In this study, we also showed that the labeling efficiency of Cu(II)-NTA to CueR_L60H_G64H mutant is above 95%. Therefore, for in cell EPR measurements, the CueR_L60H_G64H mutant was over-expressed in E. coli. The expression and purification protocols are described in the SI. SDS-PAGE confirmed CueR over-expression, and the purity of purified CueR_L60H_G64H (Fig. S1, ESI†). Circular dichroism showed that the secondary structure of the protein was not affected by this mutation (Fig. S2, ESI†). An electrophoresis mobility shift assay (EMSA) confirmed that the CueR_L60H_G64H mutant protein bound to the copA promoter in a similar manner as the native CueR protein (Fig. S3, ESI†). Assessing cell viability in the presence of Cu(II)-NTA and free Cu(II) ions at various concentrations (Fig. S4 and S5, ESI†) revealed that at the applied concentration, the cell viability is reduced by about 20%.
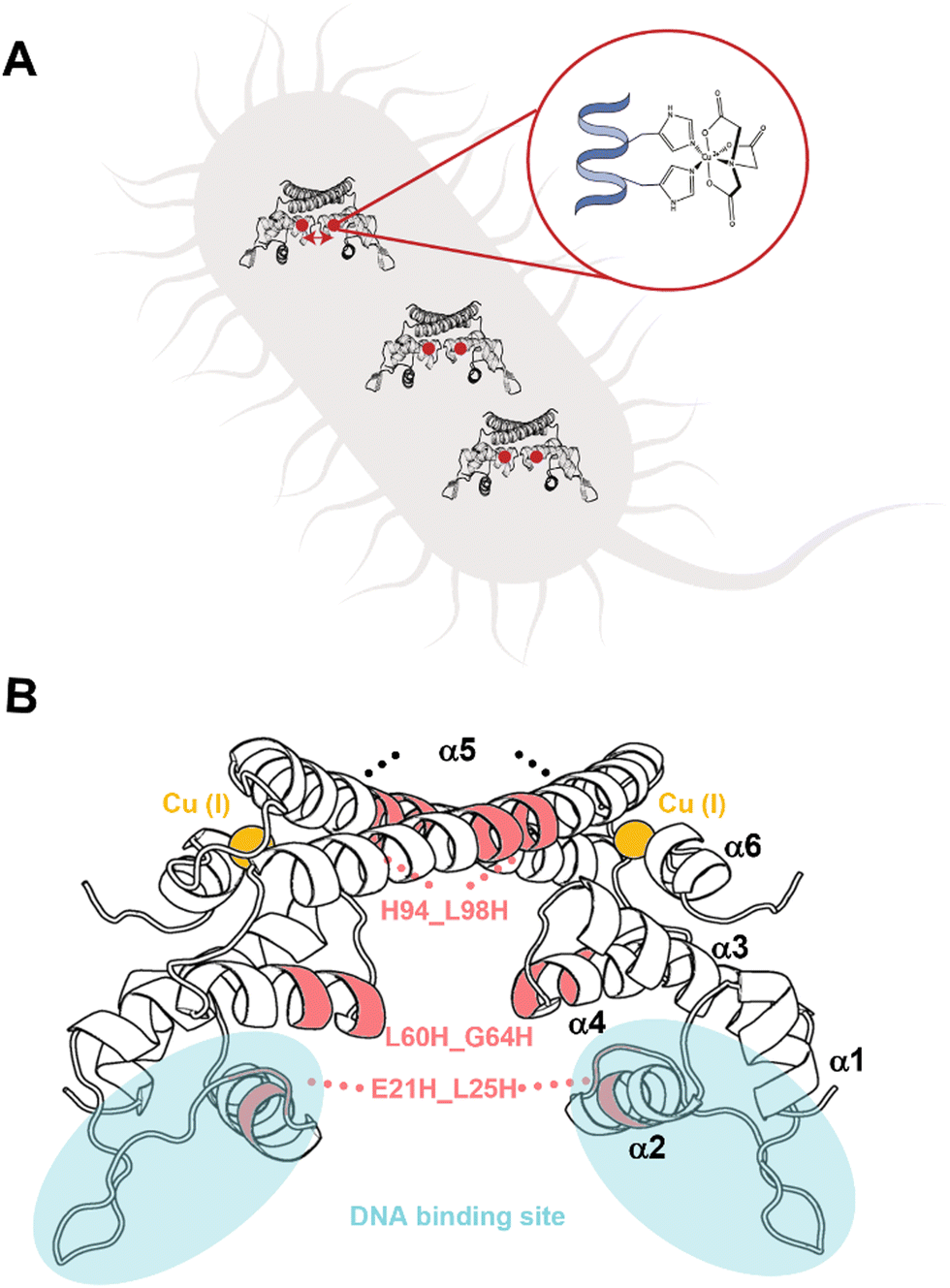 | ||
| Fig. 1 (A) Schematic depiction of the spin-labelling approach. The protein of interest is over-expressed in E. coli. It is characterized by a dHis site. Cu(II) coordinated to a nitriloacetic acid (NTA) ligand, which displays high affinity to the dHis site, is delivered into the cells to realize site-specific labelling within the cell. (B) CueR homodimer structure (PDB 1Q05). Each monomer has a ααββαααα secondary structure. α1 and α2 helices are in the DNA binding domain. The loop between α5 and α6 comprises Cu(I) site. The selected mutants are marked by pink. | ||
DEER experiments were run on three samples, namely, CueR_L60H_G64H over-expressed in E. coli cells, E. coli lysate and purified CueR_L60H_G64H (Fig. 2). The time domain DEER signals before background subtraction are presented in Fig, S6, ESI.† Cu(II)-NTA was added to the cells together with the isopropyl-β-D-thiogalactopyranoside (IPTG) inducer (first sample). To test for promoter leakage, the cells were harvested, and the cleared growth medium was measured by EPR. This revealed no Cu(II) signal (Fig. S7, ESI†). Subsequently, the harvested cells were lysed, the cell debris was collected by centrifugation, and clear lysate was measured (second sample). Finally, the data collected were compared to the purified protein (third sample). The field-sweep echo-detected EPR spectra for all three samples are shown in Fig. S8, ESI.† For in cell and lysate experiments, a Mn(II) signal was noted. However, this signal only contributes to the homogeneous background of the DEER signal and does not interfere with the DEER time domain modulations (Fig. S9, ESI†). To verify that Cu(II)-NTA cannot bind to other cellular proteins and affect the DEER signals, DEER measurements were performed on Cu(II)-NTA in E. coli cells without overexpression of CueR (Fig. S9, ESI†). The DEER signal was characterized by only exponential decay signal, confirming that there is no binding of Cu(II)-NTA to other cellular proteins. The time domain DEER signal of CueR_L60H_G64H (Fig. 2) suggested that most of the Cu(II)-NTA is bound to the protein (at least 70%). The modulation depth value of the purified CueR is 0.0048 ± 0.001, whereas CueR detected in the cell is 0.0037 ± 0.001. 70% binding allows a minimal background contribution to the DEER signal.41,42 The DEER data suggests a distance distribution for the purified protein of 2.7 ± 0.1 nm, whereas in the lysate it was 2.8 ± 0.1 nm, and 2.85 ± 0.25 nm in the cell. The predicted distance distribution based on the CueR crystal structure (PDB 1Q05) is 2.95 ± 0.1 nm. The agreement between the data confirms that most of the Cu(II)-NTA successfully bound to the over-expressed protein within the cell.
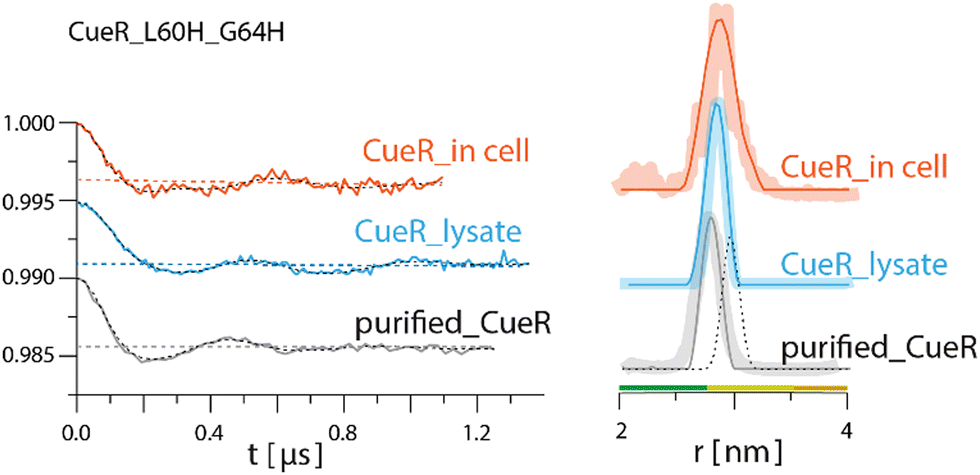 | ||
| Fig. 2 Q-band DEER measurements on CueR_L60H_G64H spin-labelled with Cu(II)-NTA. Q-band EPR distance measurements on purified (gray line) and overexpressed protein in 25 mM Tris buffer, lysate (light blue) and in BL21 cells (orange). The left side represents the time domain data after background subtraction and the right side the corresponding distance distributions. The dotted dark line represents the simulated distance distribution based on the CueR crystal structure (PDB 1Q05). Simulations were performed using the MMM program and data were analyzed using the DeerAnalysis program using Tikhonov regularization, where the regularization parameter was 50.5 Distance distribution validation considered white noise, background start and dimensionality. The colour bar indicates reliability ranges (green: shape reliable; yellow: mean and width reliable; orange: mean reliable). Purified protein concentration was 45 μM. For all samples 30% glycerol was added. | ||
To further confirm this methodology, we applied it on two additional mutants (Fig. 3), CueR_E21H_L25H, which is located at the DNA binding domain of the protein (Fig. 1B), and CueR_H94_L98H, which is located on the α5 dimerization helix. The DEER data on the purified CueR_E21H_L25H proposes a distribution of 2.7 ± 0.3 nm, while in the cell a bit narrower distribution was obtained 2.55 ± 0.1 nm. For the purified protein, additional distance distribution around 4.1 nm was observed, however this distribution is not reliable based on the distance distribution validation and the short time domain DEER signal. The 2.5–2.7 nm distributions of the purified protein and in the cell are smaller than the predicted distance distribution based on the crystal structure (PDB 1Q05). This is consistent with our previous studies, where we revealed that the two DNA binding domains are closer to each other than the crystal structure conformation and are spread apart only when bound to DNA.35,43 For the CueR_H94_L98H mutant, a distribution of 1.9 ± 0.1 nm was obtained for the purified protein, consistent with the predicted distance distribution of the crystal structure. In the cell, a distribution of around 1.8 nm was obtained. For this mutant, some longer distance distribution was observed, which might be owing to higher oligomerization state of the protein. Such oligomerization was previously detected by us when the protein concentration44,45 is high as well as by sm-FRET in cell study on CueR homologue, Zur.46 It is important to note that we did not detect major changes in the modulation depth, and the distance distribution validation suggests that the reliability of the 4.0–5.0 nm distributions is low. Therefore, we focused here only on the distribution smaller than the 2.0 nm, which agrees well with the crystal structure. For both mutants (CueR_E21H_L25H and CueR_H94_L98H), the change in the modulation depth between the purified and in cell protein was very minor, suggesting that most of the Cu(II)-NTA is bound to the protein. Moreover, the change between the distance distribution functions for all three mutants between the purified proteins and the overexpressed proteins in the cell, proposes that there are some structural differences between the cellular system and the purified proteins. These differences can be explained by the fluctuation in pH and salts present in E. coli. E. coli maintains a pH between 7.2 and 7.8 and actively transports salts in and outside of the cell, thus providing a varying environment that is different from a fixed buffer system. Secondly, CueR within the cell is surrounded by countless other macromolecules including CueR itself, which can impact the overall structure compared to the data obtained from purified proteins alone. Altogether, these minor differences from in cell data reflect the increment in understanding of structural dynamics of proteins within their physiological environment compared to in vitro data.
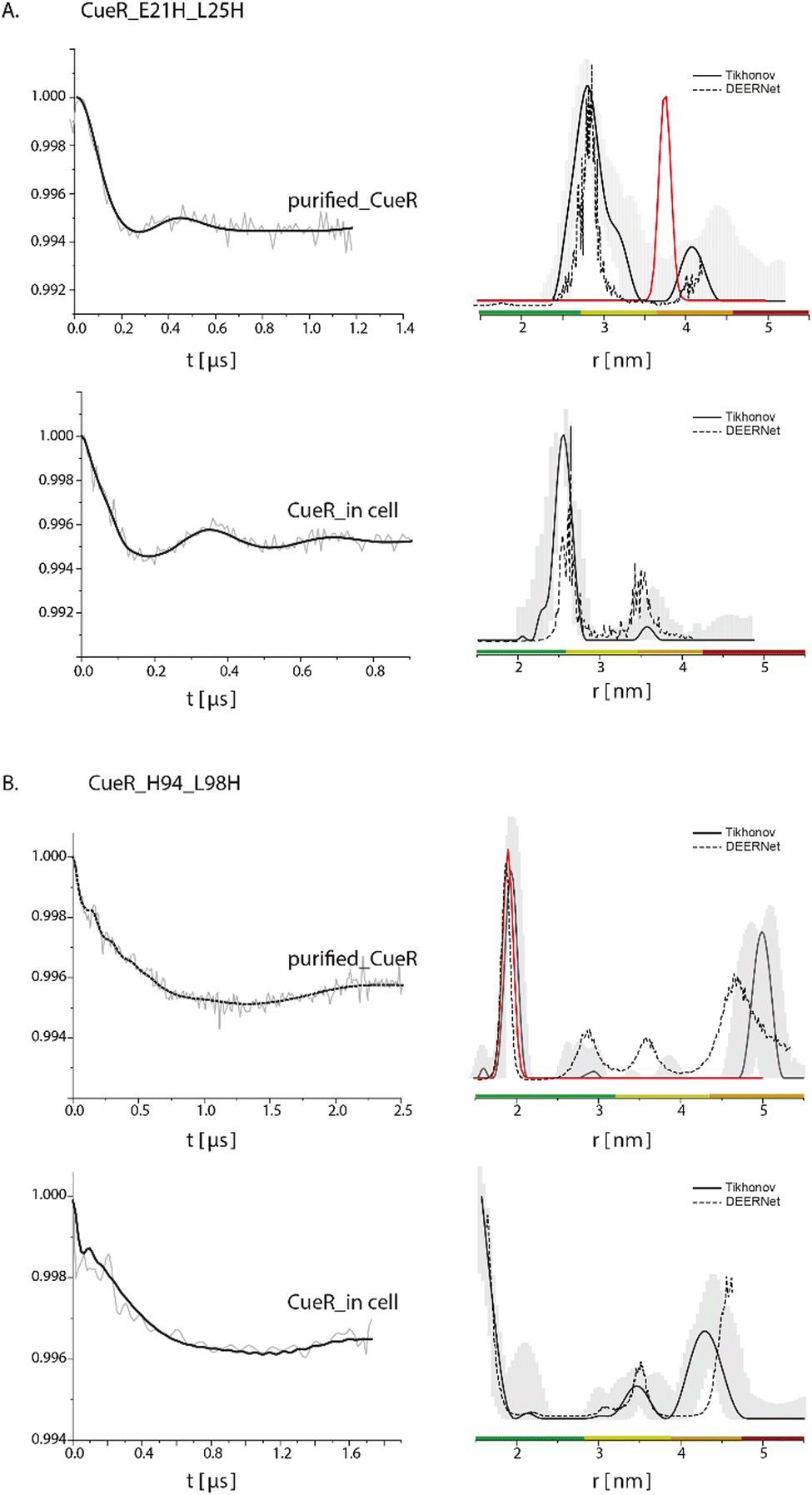 | ||
| Fig. 3 Q-band DEER measurements on (A) CueR_E21H_L25H and B. CueR_H94_L98H, spin-labelled with Cu(II)-NTA, on purified protein in 25 mM Tris buffer and in BL21 cells. The left side represent the time domain data after background subtraction and the right side the corresponding distance distributions. The red line represents the simulated distance distribution based on the CueR crystal structure (PDB 1Q05). Simulations were performed using the MMM program and data were analyzed using the DeerAnalysis program using Tikhonov regularization, where the regularization parameter was 505 (solid black lines), and using DEERNet (dashed black lines). Distance distribution validation considered white noise, background start and dimensionality. The colour bar indicates reliability ranges (green: shape reliable; yellow: mean and width reliable; orange: mean reliable; red: no quantification possible). [purified CueR_E21H_L25H] = 45 μM, [purified CueR_H94_L98H] = 100 μM. For all samples 30% glycerol was added. | ||
Gaining structural information on proteins in their native cellular environment provides novel perspectives on numerous enzymatic reaction mechanisms and facilitates development of new therapeutic approaches. During the last decade, in cell EPR spectroscopy has emerged as an excellent tool for providing high-resolution structural data on proteins. However, one of the major challenges of EPR spectroscopy is the spin-labeling process, which is mostly performed outside the cellular environment. Herein, we demonstrated the feasibility of using an endogenously over-expressed protein in E. coli with dHis site to spin-label the protein with a Cu(II) paramagnetic center in the native cellular environment. The main advantage of this method is that it can be performed on any over-expressed protein, independent of its size or complexity. The labeling yield is comparable high of at least 70%, which allows for detection of well-resolved distance distribution functions.
Y. S. and L. G. A. and M. H. performed all biochemical and EPR experiments. L. H. analyzed the data. S. R. conceived the idea, analyzed the data, and supervised the project. All authors were involved in writing the paper.
We would like to thank Dr Carmieli, Dr Seal and Prof. Goldfarb from Weizmann Institute of Science, Israel, for performing ENDOR experiments on the CueR_19F58_L60H_G64H mutant (see ESI†), and for very helpful discussions. We also acknowledge the support of the Israel Science Foundation (grant 176/16 and 212/22).
Conflicts of interest
There are no conflicts to declare.References
- E. Luchinat and L. Banci, Curr. Opin. Struct. Biol., 2022, 74, 102374 CrossRef CAS PubMed.
- E. Luchinat, M. Cremonini and L. Banci, Chem. Rev., 2022, 122, 9267–9306 CrossRef CAS PubMed.
- H. E. Grecco and P. J. Verveer, Chem. Phys. Chem., 2011, 12, 484–490 CrossRef CAS PubMed.
- A. Pietraszewska-Bogiel and T. W. Gadella, J. Microsc., 2011, 241, 111–118 CrossRef CAS PubMed.
- G. Jeschke, V. Chechik, P. Ionita, A. Godt, H. Zimmermann, J. Banham, C. R. Timmel, D. Hilger and H. Jung, Appl. Magn. Reson., 2006, 30, 473–498 CrossRef CAS.
- V. Lucic, A. Leis and W. Baumeister, Cell Biol, 2008, 130, 185–196 CAS.
- E. Callaway, Revolutionary Nat., 2020, 578, 201 CAS.
- W. Baumeister, Biochem. Biophys. Res. Commun., 2022, 633, 26–28 CrossRef CAS PubMed.
- D. Goldfarb, Curr. Opin. Struct. Biol., 2022, 75, 102398 CrossRef CAS PubMed.
- S. L. Meichsner, Y. Kutin and M. Kasanmascheff, Angew. Chem., Int. Ed., 2021, 60, 19155–19161 CrossRef CAS PubMed.
- S. Kucher, S. Korneev, J. P. Klare, D. Klose and H. J. Steinhoff, Phys. Chem. Chem. Phys., 2020, 22, 13358–13362 RSC.
- Y. Yang, F. Yang, X. Y. Li, X. C. Su and D. Goldfarb, J. Phys. Chem. B, 2019, 123, 1050–1059 CrossRef CAS PubMed.
- L. John and M. Drescher, Bio-Protoc., 2018, 8, e2798 Search PubMed.
- M. Qi, A. Gross, G. Jeschke, A. Godt and M. Drescher, J. Am. Chem. Soc., 2014, 136, 15366–15378 CrossRef CAS PubMed.
- M. Azarkh, O. Okle, P. Eyring, D. R. Dietrich and M. Drescher, J. Magn. Reson., 2011, 212, 450–454 CrossRef CAS PubMed.
- M. J. Schmidt, J. Borbas, M. Drescher and D. Summerer, J. Am. Chem. Soc., 2014, 136, 1238–1241 CrossRef CAS PubMed.
- O. Schiemann, et al. , J. Am. Chem. Soc., 2021, 143, 17875–17890 CrossRef CAS PubMed.
- K. Ackermann, J. L. Wort and B. E. Bode, J. Phys. Chem. B, 2021, 125, 5358–5364 CrossRef CAS PubMed.
- Y. Yang, F. Yang, Y. J. Gong, T. Bahrenberg, A. Feintuch, X. C. Su and D. Goldfarb, J. Phys. Chem. Lett., 2018, 9, 6119–6123 CrossRef CAS PubMed.
- A. Bonucci, O. Ouari, B. Guigliarelli, V. Belle and E. Mileo, ChemBioChem, 2020, 21, 451–460 CrossRef CAS PubMed.
- P. Widder, J. Schuck, D. Summerer and M. Drescher, Phys. Chem. Chem. Phys., 2020, 22, 4875–4879 RSC.
- N. Fleck, C. A. Heubach, T. Hett, F. R. Haege, P. P. Bawol, H. Baltruschat and O. Schiemann, SLIM: A Short-Linked, Angew. Chem., Int. Ed., 2020, 59, 9767–9772 CrossRef CAS PubMed.
- V. M. Tormyshev, A. S. Chubarov, O. A. Krumkacheva, D. V. Trukhin, O. Y. Rogozhnikova, A. S. Spitsyna, A. A. Kuzhelev, V. V. Koval, M. V. Fedin, T. S. Godovikova, M. K. Bowman and E. G. Bagryanskaya, Chemistry, 2020, 26, 2705–2712 CrossRef CAS PubMed.
- S. Jana, E. G. B. Evans, H. S. Jang, S. Zhang, H. Zhang, A. Rajca, S. E. Gordon, W. N. Zagotta, S. Stoll and R. A. Mehl, J. Am. Chem. Soc., 2023, 145(27), 14608–14620 CrossRef CAS PubMed.
- A. Gamble Jarvi, X. Bogetti, K. Singewald, S. Ghosh and S. Saxena, Acc. Chem. Res., 2021, 54, 1481–1491 CrossRef CAS PubMed.
- J. L. Wort, K. Ackermann, D. G. Norman and B. E. Bode, Phys. Chem. Chem. Phys., 2021, 23, 3810–3819 RSC.
- S. Ghosh, M. J. Lawless, G. S. Rule and S. Saxena, J. Magn. Reson., 2018, 286, 163–171 CrossRef CAS PubMed.
- A. Meir, G. Walke, F. Schwerdtfeger, L. Gevorkyan Airapetov and S. Ruthstein, PLoS One, 2019, 14, e0219337 CrossRef CAS PubMed.
- A. Magistrato, M. Pavlin, Z. Qasem and S. Ruthstein, Curr. Opin. Struct. Biol., 2019, 58, 26–33 CrossRef CAS PubMed.
- Y. Yoshida, S. Furuta and E. Niki, Biochim. Biophys. Acta, 1993, 1210, 81–88 CrossRef CAS PubMed.
- T. F. Cunningham, M. R. Putterman, A. Desai, W. S. Horne and S. Saxena, Angew. Chem., Int. Ed., 2015, 54, 6330–6334 CrossRef CAS PubMed.
- L. Pauling, R. B. Corey and H. R. Branson, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 205–211 CrossRef CAS PubMed.
- C. Fang, S. J. Philips, X. Wu, K. Chen, J. Shi, L. Shen, J. Xu, Y. Feng, T. V. O'Halloran and Y. Zhang, Nat. Chem. Biol., 2021, 17, 57–64 CrossRef CAS PubMed.
- H. Sameach, S. Ghosh, L. Gevorkyan-Airapetov, S. Saxena and S. Ruthstein, Angew. Chem., Int. Ed., 2019, 58, 3053–3056 CrossRef CAS PubMed.
- H. Sameach, A. Narunsky, S. Azoulay-Ginsburg, L. Gevorkyan-Aiapetov, Y. Zehavi, Y. Moskovitz, T. Juven-Gershon, N. Ben-Tal and S. Ruthstein, Structure, 2017, 25, 988–996 CrossRef CAS PubMed e983.
- D. J. Martell, C. P. Joshi, A. Gaballa, A. G. Santiago, T. Y. Chen, W. Jung, J. D. Helmann and P. Chen, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13467–13472 CrossRef CAS PubMed.
- K. Chen, S. Yuldasheva, J. E. Penner-Hahn and T. V. O'Halloran, J. Am. Chem. Soc., 2003, 125, 12088–12089 CrossRef CAS PubMed.
- A. Changela, K. Chen, Y. Xue, J. Holschen, C. E. Outten, T. V. O'Halloran and A. Mondragon, Science, 2003, 301, 1383–1387 CrossRef CAS PubMed.
- F. W. Outten, C. E. Outten, J. Hale and T. V. O'Halloran, J. Biol. Chem., 2000, 275, 31024–31029 CrossRef CAS PubMed.
- I. Yakobov, A. Mandato, L. Hofmann, K. Singewald, Y. Shenberger, L. Gevorkyan-Airapetov, S. Saxena and S. Ruthstein, Protein Sci., 2022, 31, e4309 CrossRef CAS PubMed.
- S. Ketter, A. Gopinath, O. Rogozhnikova, D. Trukhin, V. M. Tormyshev, E. G. Bagryanskaya and B. Joseph, Chemistry, 2021, 27, 2299–2304 CrossRef CAS PubMed.
- S. F. Haysom, J. Machin, J. M. Whitehouse, J. E. Horne, K. Fenn, Y. Ma, H. El Mkami, N. Bohringer, T. F. Schaberle, N. A. Ranson, S. E. Radford and C. Pliotas, Angew. Chem., Int. Ed., 2023, e202218783 Search PubMed.
- R. Schwartz, S. Ruthstein and D. T. Major, J. Phys. Chem. B, 2021, 125, 9417–9425 CrossRef CAS PubMed.
- J. Casto, A. Mandato, L. Hofmann, I. Yakobov, S. Ghosh, S. Ruthstein and S. Saxena, Chem. Sci., 2022, 13, 1693–1697 RSC.
- M. Pavlin, Z. Qasem, H. Sameach, L. Gevorkyan-Airapetov, I. Ritacco, S. Ruthstein and A. Magistrato, Int. J. Mol. Sci., 2019, 20(14), 3462 CrossRef CAS PubMed.
- W. Jung, K. Sengupta, B. M. Wendel, J. D. Helmann and P. Chen, Nucleic Acids Res., 2020, 48, 2199–2208 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The materials and methods as well as additional control experiments. See DOI: https://doi.org/10.1039/d3cc03047d |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |