
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of regiodivergent asymmetric reductive coupling reactions of allenamides to access heteroatom-rich organic compounds
Stephen
Collins
and
Joshua D.
Sieber
 *
*
Virginia Commonwealth University, Department of Chemistry 1001 West Main Street, Richmond, VA 23284, USA. E-mail: jdsieber@vcu.edu
First published on 27th July 2023
Abstract
Organic compounds of biological importance often contain multiple stereogenic C-heteroatom functional groups (e.g. amines, alcohols, and ethers). As a result, synthetic methods to access such compounds in a reliable and stereoselective fashion are important. In this feature article, we present a strategy to enable the introduction of multiple C-heteroatom functional groups in a regiodivergent cross-coupling approach through the use of reductive coupling chemistry employing allenamides. Such processes allow for opportunities to access different heteroatom substitution patterns from the same starting materials.
 Stephen Collins | Stephen Collins received his B.S. degree in chemistry from the University of North Carolina at Charlotte while working under Prof. Christopher Bejger. He is currently a PhD student in the Chemistry Department at the Virginia Commonwealth University. His doctoral studies are focused on asymmetric Cu-catalysed reductive coupling reactions utilizing allenamides to access chiral diamines and aminoalcohols. |
 Joshua D. Sieber | Joshua Sieber is an Assistant Professor in the Chemistry Department at the Virginia Commonwealth University since 2018. Prior to his academic appointment, he was a senior (2011 – 2016) and principal (2016 – 2018) scientist at Boehringer Ingelheim Pharmaceuticals, Inc. in the Chemical Development Process Chemistry Group. Having a background in complex organic molecule synthesis and asymmetric catalysis, his current research interests focus on developing catalytic asymmetric C–C bond forming reactions. |
Introduction
When considering organic architectures of important biological and medicinal activity, such compounds are often found to contain multiple heteroatom functional groups in a chiral arrangement (Fig. 1). As an example, the 1,2-aminoalcohol1,2 motif alone is present in >300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds, >2000 natural products, and >80 Food and Drug Administration-approved drugs, according to a survey from 2019.3 As a result, methodologies and synthetic strategies to enable efficient synthetic access to these useful “heteroatom-rich” organic compounds is an important research endeavour.
000 compounds, >2000 natural products, and >80 Food and Drug Administration-approved drugs, according to a survey from 2019.3 As a result, methodologies and synthetic strategies to enable efficient synthetic access to these useful “heteroatom-rich” organic compounds is an important research endeavour.
 | ||
| Fig. 1 Selected heteroatom-rich (i.e. N, O) natural products and bioactive organic compounds. | ||
In the context of identifying new tools to allow for the programmatic synthesis of diverse heteroatom-rich organic compounds, it can be argued that an ideal4,5 strategic approach to access a functional group pair could arise through the use of a convergent6–8 cross-coupling type process (Fig. 2). For example, catalytic coupling of two functionalized fragments X and Y enables access to a bis(functionalized) product through forging of the C–C bond (Fig. 2A). According to this design strategy, it is expected that the specific electronic nature of the functional groups present within X and Y (i.e. FG1 and FG2) will dictate the type of cross-coupling reaction required to achieve the desired bond formation (Fig. 2B–D).9–11 For instance, for a case where one coupling fragment contains and electron-withdrawing functional group (EWG) and the other contains an electron-donating functional group (EDG), the carbon atoms attached to each functional group are polarized to be electrophilic and nucleophilic, respectively (Fig. 2B).9–11 This scenario creates a common reactivity profile observed for two-electron chemical processes where each fragment contains a “matched” polarity profile leading to reagents that are poised to react with one another through a redox neutral process (electrophile/nucleophile pair). Classical cross-coupling processes12 proceed by this type of redox neutral process (e.g. Suzuki–Miyaura,13 Negishi,14 Sonogashira,15etc.). However, for the case of fragments bearing two electron-donating functional groups (Fig. 2C: 4,5), these two nucleophilic fragments9,10 result in an excess amount of electrons needed to forge the desired product 6, and therefore, a stoichiometric oxidant is required to absorb the excess electrons in an oxidative coupling process.16,17 In the last option, coupling partners that both contain electron-withdrawing functional groups (7,8; Fig. 2D), leads to a scenario whereby a deficiency of electrons needed to complete the desired coupling exists and as a result, a stoichiometric reductant is necessary in an overall reductive process.18,19
 | ||
| Fig. 2 Idealized strategy for the convergent integration of multiple functional groups through a convergent cross-coupling approach. EDG = electron-donating group, EWG = electron-withdrawing group. | ||
In relation to applying the cross-coupling approach outlined in Fig. 2 to the synthesis of N- and O-containing heteroatom-rich organic compounds (Fig. 1), a reductive coupling process would be needed considering the electron-withdrawing nature of N- and O-based substituents (Fig. 2D). Catalytic reductive coupling processes are known to occur through two types of mechanistic scenarios based on the nature of the reducing agent (Fig. 3A).18 Pioneering work in the area of metal-catalysed reductive coupling began to emerge in the mid-late 1990's through the work of Mori,20 Tamaru,21 Montgomery,22 Jamison,23 Buchwald,24 Kang,25 and Kurosawa26 through the Ni-catalysed coupling of unsaturated hydrocarbons with aldehydes27 utilizing a terminal reductant to enable catalyst turnover through incorporation of atoms from the reductant into the final product (Type 1 process). Since that time, numerous researchers have developed powerful C–C bond forming processes based on this approach using a variety of different atom-transfer reductants (e.g. HSiR3, ZnR2, B2(OR)4, etc).18,19 In the late 2000s a resurgence in reductive coupling reaction development began for initially cross-electrophile sp2–sp3 coupling reactions of aryl and alkyl halides by the groups of Lipshutz,28 Wangelin,29,30 Gong31 and Weix.32,33 These processes may proceed either by in situ formation of an organometallic nucleophile followed by a classical redox neutral coupling,34 or in the case of Ni-catalysis, through a Type 2 reductive coupling process requiring electron-transfer from the reductant to the catalyst to enable catalyst turnover.33 Such Type 2 techniques have been extended by a variety of researchers to other cross-electrophile couplings beyond aryl/alkyl sp2–sp3 couplings with the requisite electrons provided from either stoichiometric transition metal reductants (e.g. Zn or Mn),35,36 photocatalysis,37,38 or electrochemically.39–42
 | ||
| Fig. 3 Considerations for metal-catalysed reductive coupling as a tool for multiple heteroatom incorporation. | ||
It should be pointed out that an additional opportunity to the strategy outlined in Fig. 2D can be realized when considering the entire carbon framework of one of the coupling partners (e.g.10, Fig. 3B). For instance, in the simplified coupling to access 9, only the 1,2-substituted product from C1-coupling is formed; however, in the case of 10, if site-selective catalytic coupling with 7 could be realized through modification of the catalyst and reaction conditions, then a variety of heteroatom-substitution patterns (i.e.11–13) may be accessed from the same starting materials. Along these lines, processes affording coupling at C1 and C3 of 10 would be required to be reductive in nature due to the chain polarization of 10 by the electron-withdrawing group, whereas coupling at C2 may be redox neutral. These electronic differences may be utilized to aid in the development of such site-selective, regiodivergent reaction processes.
The Sieber research group has been interested in developing methodologies for the asymmetric and regiodivergent reductive coupling of N- and O-substituted coupling partners according to the outlined strategy given in Fig. 3B as a tool for the synthesis of heteroatom-rich chiral organic compounds (Fig. 1).43–50 As a result, we have primarily focused on accessing coupling products at C1 and C3 of the hypothetical starting material 10 (e.g.11 and 13, respectively) since these products are typically more challenging to access due to the dissonant charge affinity9–11 produced by the relationship of the two electron-withdrawing heteroatom substituents.
To realize the outlined strategy of Fig. 3B, an appropriate reagent for 10 is necessary that can enable regiodivergent access to products such as 11 and 13. We envisioned this could be achieved using a N-substituted allene reagent (16, Fig. 4)51,52 under a Type 1 reductive allylative process.53 For instance, sigma-bond metathesis of a M-OR complex (14) with reductant X–Y followed by metalation of 16 may provide an equilibrating mixture of nucleophilic substituted allyl metal complexes l-17 and b-17. Addition of these reagents to carbonyl or imine electrophiles (18) through chair-like transition states would provide access to either 1,2- (19) or 1,4-heteroatom (20) substituted products, respectively. Reductive allylation techniques incorporating a hydrogen atom from the reductant X–Y have been largely pioneered by the Krische group18 utilizing allenes,54–57 1,3-dienes,58–60 or enynes.61–63 These powerful reactions represent atom economical alternatives to classical allylation processes utilizing preformed organometallic reagents and are typically catalysed by either Rh, Ir, or Ru. More recently, the Buchwald group64–68 has developed orthogonal reductive allylation reactions under Cu-catalysis using silane as the terminal reductant. However, all these past precedented processes are typically branched-selective and access to the linear products are rare.43,47,69 Furthermore, much of the work in this area has investigated the use of unsaturated hydrocarbon coupling partners bearing only carbon-substituents and the application and use of N-substituted reagents (e.g.16) has been less well developed prior to our work.3,54,70,71
 | ||
| Fig. 4 Proposed reaction designs for multi-heteroatom functional groups. | ||
In order to enable regiodivergency in reductive allylation reactions of 16, our design strategy proposes incorporation of a tethered coordinating group ‘G’ in 16 as a technique to enable generation of the less common linear product 20 in these reactions. It was proposed that using a catalyst with a low coordination number (e.g. use of monodentate ligands) at the metal may facilitate formation of 20 through stabilization of b-17 through internal coordination of the directing group ‘G’. In contrast, use of a coordinatively saturated metal catalyst may in turn restore the more commonly observed branched-selectivity by inhibition of G-group binding to furnish 19. As a result, access to highly valuable chiral 1,2-aminoalcohols and diamines or γ-lactones or lactams may be accessible from the same starting materials through selection of the reaction conditions. The development of these types of asymmetric strategies utilizing reagents 16 are summarized herein.
Asymmetric auxiliary-controlled regiodivergent Cu-catalysed reductive coupling of allenamides and ketones
To realize the proposed strategy outlined in Fig. 4, an appropriate structure for 16 was required. Our group initially chose to investigate the use of the Evans-oxazolidinone derived allenamide 21a (Scheme 1) for these processes that has been pioneered by the Hsung group51 for other non-reductive coupling reactions due to the low cost of the Evans auxiliary.45 Accordingly, the carbonyl group of the oxazolidinone must serve as the G-group of 16 to enable linear control. Furthermore, we chose to study Cu-catalysed reductive coupling processes due to the improved sustainability and safety of Cu relative to the competitive Type 1 reductive coupling catalysts derived from the precious metals Ru, Rh, or Ir due to the high availability, low cost, and reduced toxicity of Cu compared with these other precious metal pre-catalysts.50 Gratifyingly, a systematic study of monodentate phosphorous based ligands in the coupling of 21a with ketone electrophiles demonstrated that high linear selectivity could be obtained to provide homoaldol products 22 in excellent yield and stereocontrol when using phosphoramidite ligand (PhO)2PNMe2.43 The substrate scope in this process was shown to be quite broad, and an average stereocontrol of 83:17 dr could be obtained for the 21 examples investigated. A more detailed investigation47 into factors controlling diastereoselectivity then led to the identification of an alternative system employing chiral allenamide 21b with a readily available commercial aryl phosphite ligand to provide improved stereocontrol across the same 21 examples (93:7 avg. dr). Furthermore, empirical ligand studies supported the proposal that oxazolidinone coordination to Cu (e.g. b-24) enables linear control based on the observation that linear selectivity increased with decreasing ligand electron-donating ability (i.e. increasing Lewis acidity at Cu) and bidentate phosphine ligands led to a switch to branched selectivity.43,44 Additionally, the observed product stereochemical outcome (Z-olefin geometry, alcohol absolute configuration) further supports oxazolidinone binding as an Evans chelate model72,73 chair-like transition structure TS-1 correctly rationalizes the observed stereochemistry.
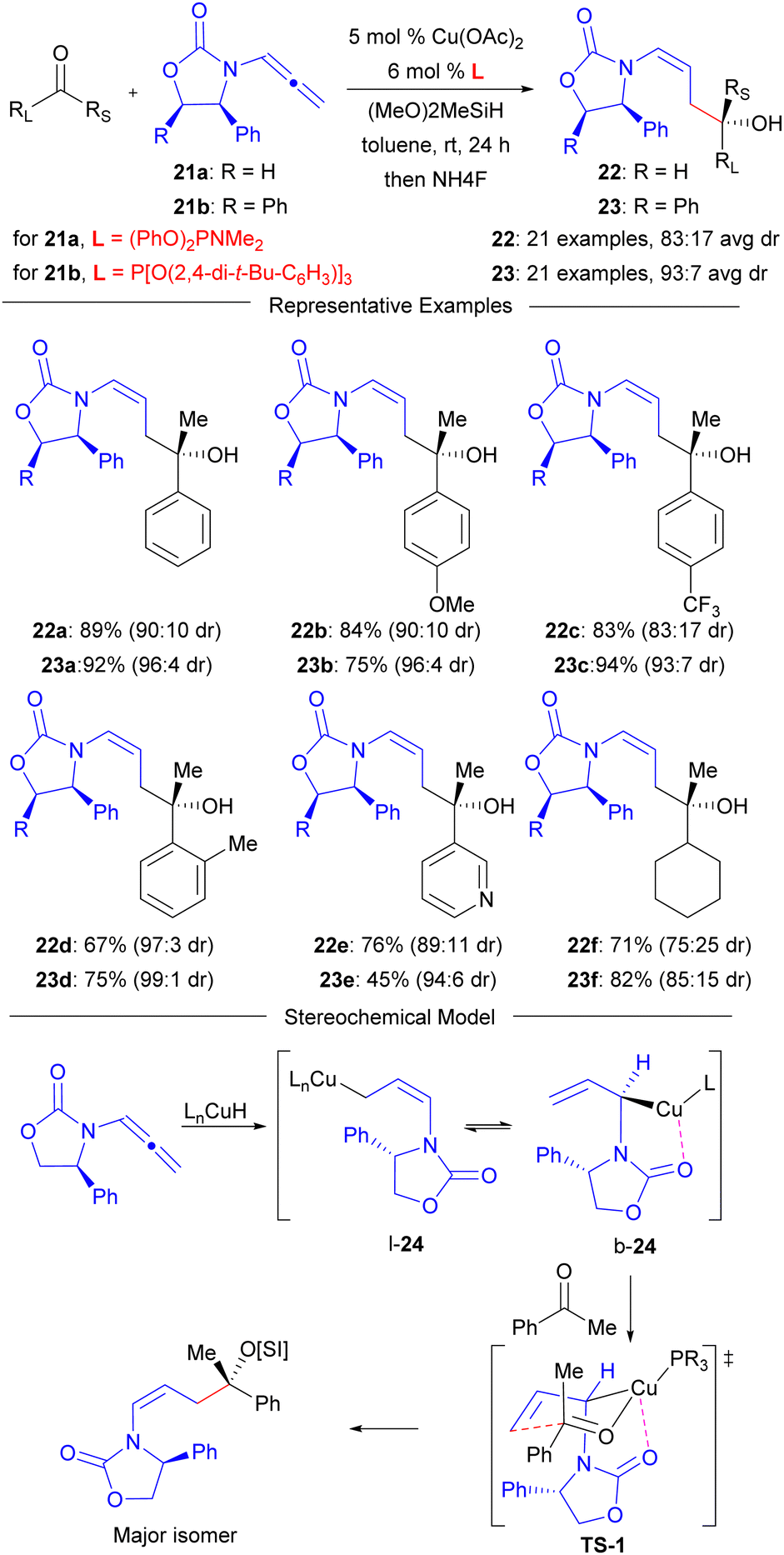 | ||
| Scheme 1 Linear-selective reductive coupling of chiral oxazolidinone derived allenamides and ketones. | ||
Upon obtaining confidence in our proposed design for regiocontrol, we next turned to developing a branched selective protocol through inhibition of oxazolidinone complexation to the Cu-catalyst.44 While bidentate ligands were found to provide a turnover from linear to branched selectivity, ultimately use of the sterically hindered and highly electron-rich N-heterocyclic carbene (NHC) family of ligands proved optimal (Scheme 2). While this system was shown to be more sensitive to steric effects than the linear-selective system (Scheme 1), it was still able to produce branched products with good yields and high regio- and stereoselectivities. The ability of the NHC-based ligands to provide high branched regioselectivity is consistent with their well-known strong electron-donating74 ability that may reduce the Lewis acidity at the Cu-catalyst to inhibit oxazolidinone binding. However, the steric effects induced at the metal catalyst by an NHC ligands is expected to be quite different relative to the cone-type effects of PR3 type ligands measured by the cone angle.75 Steric shielding of the Cu-catalyst towards oxazolidinone complexation by the mesityl-groups is also possibly responsible for the switch to branched selectivity. This possibility was corroborated by the observation that improved branched selectivity was observed when switching to the NHC ligand SIPr having a more sterically encumbered 2,6-di-isopropylphenyl N-substituent on the NHC.44 Finally, the observed major diastereomer produced in the reaction could be rationalized by an Evans dipole-minimization model (TS-2).72,76
 | ||
| Scheme 2 Branched-selective reductive coupling of a chiral oxazolidinone derived allenamide and ketones. | ||
The synthetic utility of the linear and branched reductive coupling products generated from the regiodivergent Cu-catalysed methodology was demonstrated through removal of the Evans oxazolidinone auxiliary (Scheme 3). Overall, the linear-selective reaction enables access to homoaldol synthons (22). Homoaldol derivatives are typically prepared through the generation of homoenolates,77,78 and asymmetric variants are minimal.73,79,80 Linear-product 22g underwent iodocyclization using NIS followed by an E1cb-elimination through Li-halogen exchange with n-BuLi to provide dihydrofuran 27 in excellent yield along with 86% recovery of the Evans oxazolidinone.47 Dihydrofurans are useful synthetic intermediates that can be directly converted to γ-lactones through PCC oxidation81 that enabled the synthesis of the natural product (S)-(−)-boivinianin82 A. In regards to branched products 25, notably these were highly crystalline and could be enriched to single diastereomers using crystallization from hexanes/ethyl acetate mixtures.44 Base- induced carbonate migration allowed access to 28 whereby the phenethanol motif can be cleaved using a telescoped alcohol elimination process followed by enamide hydrolysis to produce 29 that can be hydrolysed to the 1,2-aminoalcohol if desired.
 | ||
| Scheme 3 Auxiliary removal. | ||
Overall, the methodologies outlined in Schemes 1 and 2 enable regiodivergent aminoallylation processes through generation of the reactive allylic organometallic nucleophile in a catalytic fashion under ambient conditions. Prior access to such compounds classically relied on generation of preformed organometallic nucleophiles from aza-allylanions (Scheme 4).73,83–86 The paradigm shift in recent trends for carbonyl allylation employing allylmetal generation in situ by a metal catalyst has expanded functional group compatibility in these processes since strong bases and cryogenic temperatures are no longer required.
 | ||
| Scheme 4 Classical Aminoallylation Methods. | ||
Asymmetric auxiliary-controlled Cu-catalysed reductive coupling of allenamides and imines: 1,2-diamine synthesis
After establishing the reductive coupling reactions utilizing ketone electrophiles, imines were next investigated as a protocol to access chiral 1,2-diamines (Scheme 5). Here, the monodentate electron-rich phosphine ligand PCy3 was identified as optimal, and it was discovered that this system could provide access to both chiral urea or diamine synthons depending on the introduction of tert-butanol as an additive.45 The use of alcohol additives has been well established for CuH catalysed reductive coupling reactions.87,88 Mechanistically, it was proposed that these followed a process similar to that involving the branched 1,2-aminoalcohol, wherein linear allylcopper reagent 43 reacted with aldimine 40 through a chair-like transition structure to produce intermediate 44. In the ketone variant of these reactions (vide infra), the analogous O-intermediate of 44 would turnover through direct silylation; however, due to the weak bond strength of the N–Si bond,89 there is no driving force for direct turnover through silylation of 44. As a result, in the absence of tert-butanol, intramolecular carbamate migration of 44 can occur to afford Cu-alkoxide intermediate 45 that can turnover through direct silylation leading to urea product 46. In contrast, in the presence of t-butanol, direct protonolysis of 44 can occur to generate copper alkoxide intermediate LnCuOtBu that can regenerate the CuH catalyst by silylation with silane. | ||
| Scheme 5 Branched-selective reductive coupling of a chiral oxazolidinone derived allenamide and imines. | ||
In the context of the substrate scope for the reaction with imine electrophiles, both electron-rich (40b,c) and electron-poor (40a,d,e,f) aryl aldimines participated well to afford the desired 1,2-diamine derivatives in high yields as single diastereomers.45 Sterically hindered ortho-substituted aryl aldimines (40c) were also suitable coupling partners when the aldimine N-DMB protecting group was replaced with a less hindered N-benzyl group along with an increase in reaction temperature. Detailed density functional theory calculations on the reaction system were performed and revealed that stereo- and regiocontrol in the process was a result of Curtin–Hammett kinetics where a rapidly equilibrating mixture of N-substituted allylic Cu nucleophiles were present with the major product resulting from the lowest energy transition state for the addition of these rapidly equilibrating nucleophiles to the electrophilic imine through chair-like transition structures. Unfortunately, in regards to a regiodivergent process, the linear isomer of product was not observed under a variety of conditions tested. This is likely due to inhibition of the chair-like transition structure needed to access the linear product due to additional axial interactions between the R2-group of the imine with the oxazolidinone that is not present when ketone electrophiles are used (i.e.TS-1, Scheme 1).
The potential synthetic utility of the imine reductive coupling methodology was applied to synthesis of 48 as a potential surrogate to the potent NK-1 inhibitor CP-9999490 (Scheme 6). The Cu-catalysed reductive coupling reaction was demonstrated on a 1.0 gram-scale on the benchtop using standard Schlenk techniques in excellent yield to provide 41g as a single diastereomer. Subsequent allylation of the PMB amine followed by ring-closing metathesis afforded 48.
 | ||
| Scheme 6 Advancement to a cyclic diamine synthon. | ||
The Prasad group91 recently investigated the use of the aza-allyl anion produced by deprotonation of 30 with strong base in the addition reactions with Ellman's auxiliary92 derived chiral sulfinyl imines (Scheme 7). The regiocontrol was determined to be dependent on the nature of the R1-group of the sulfinyl imine. In general, branched products 50 were favoured, however, as the steric size of the R1-group increases, the amount of linear product 51 increases. The highest yields of linear proudcts 51 was obtained for ortho-substituted aryl R1-groups. In general, excellent diasteroselectivities were obtained in these processes, but the identity of the major diastereomer of 51 was not determined.
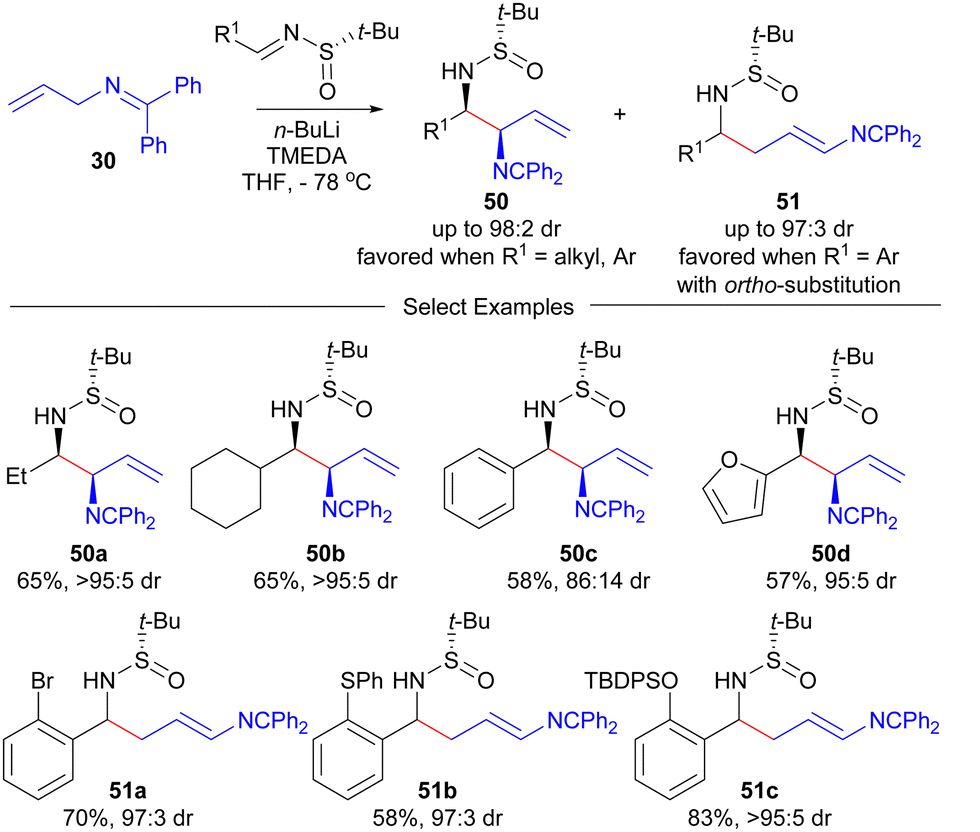 | ||
| Scheme 7 Prasad's aza-allyl anion based addition to Ellman's auxiliary derived sulfinyl imines. | ||
Enantioselective Cu-catalysed reductive coupling of allenamides and carbonyl-based electrophiles: 1,2-aminoalcohol synthesis
While the initial studies confirmed the ability to access alternate heteroatom substitution patterns from the same allenamide starting material through catalyst modulation,43,44,47 stereocontrol was achieved by the chirality of the allenamide. It is reasonable to consider that an appropriately tuned chiral Cu-catalyst could enable the same type of transformations through enantioselective catalysis while utilizing an achiral allenamide. Such a strategy is attractive as the necessary chiral information can be used in sub-stoichiometric quantities. Towards this goal, initial investigations into the application of a simple achiral allenamide analogue to the past Evans’ auxiliary derived system (i.e.52) using chiral bis(phosphine) ligands to generate branched reaction product (53) led to the identification of Walphos-8 (W8) as optimal in reactions utilizing aromatic ketones (Scheme 8).46 Notably, it was observed that carbamate rearrangement product 54 was obtained as a side-product in these processes in variable amounts as a function of the nature of the R1-alkyl group of the ketone. For instance, as the size of the R1-group increased (i.e. Me to Et), the amount of rearrangement product 54 also increased. Furthermore, it was initially surprising to note that the enantiopurities of 53 and 54 were not identical. These results implied that a reversible allylcupration event (56 → 57) was occurring along with an on-cycle rearrangement process to 54 (57 → 58 → 59 → 54·Si). Because the intermediate stereoisomers of 57 do not need to convert to 54·Si at the same rate, epimerization of 57 can occur throughout the carbamate migration event due to the reversible allylcupration step. This competition in catalytic turnover events also rationalizes the effect of R1-group on product distribution. As the R1-group becomes larger, the rate of silylation of 57 (a bimolecular process) is decreased whereas the rate of carbamate rearrangement (a unimolecular process) is increased due to an enhanced Thorpe-Ingold effect93,94 leading to increased amounts of rearranged product 54. Further support for the existence of a reversible allylcupration step was provided through reaction of 60, whose nitrogen was masked by a group that could not undergo rearrangement, with a Walphos-8 derived Cu-alkoxide catalyst as a way to regenerate an analogue to intermediate 57 that may not rearrange (i.e.61). Retroallylation95 was observed to occur from this intermediate by observation of the formation of ketone (28%) and protonolysis products 63 and 64. This is the first example of reversible allylation in metal-catalysed reductive coupling reactions. | ||
| Scheme 8 Enantioselective reductive coupling of ketones and a cyclic allenamide to 1,2-aminoalcohol synthons. | ||
The overall reaction scope to access the syn-1,2-aminoalcohol synthons 53 was examined using the Walphos-8 derived Cu-catalyst system (Scheme 9).46 Good yields with variable enantioselectivities were obtained for a variety of ketones when using a direct silylative turnover mechanism (Method A). The variable enantiocontrol was not surprising due to the determined on-cycle carbamate rearrangement problem (Scheme 8), and it was reasoned that additives that may facilitate the trapping of intermediate 57 may improve enantiocontrol. As such, the addition of t-butanol was developed under the idea that direct protonolysis of 57 may improve enantiocontrol (protonolysis turnover mechanism, Method B). In most cases, this resulted in an increase in enantiopurity but a decrease in yield was also obtained due to competitive protonation of the intermediate substituted allylic copper nucleophile (i.e.56). Notably, the ligand used in these processes can also be readily recovered which was demonstrated on a 1.0 mmol scale reaction.
 | ||
| Scheme 9 Ketone scope in the (Walphos-8)Cu-catalysed reductive coupling of ketones and a cyclic allenamide. | ||
Due to the variable enantioselectivities obtained when using cyclic carbamate derived allenamides that can undergo competitive on-cycle carbamate migration (e.g.52), it was hypothesized that substrate generality could be improved by using acyclic allenamides bearing substituents that could not undergo this problematic rearrangement (e.g. N-Ac, N-Bz, etc.). As a result, a second-generation system was investigated under this context and an optimal enantioselective system was identified when employing acyclic allenamide 66 in conjunction with a Josiphos-2 (J2) derived chiral Cu-catalyst (Scheme 10).48 Notably, this new system was found to produce the opposite diastereomer in the reaction as compared to allenamide 52 (anti-selectivity obtained), and the yields and enantioselectivities for both electron-withdrawing and electron-donating substituted aryl ketones were high. Excellent results were also obtained for heterocycles (67c,f). Removal of the N-acetyl moiety of 67 could be achieved using lithium amidotrihydroborate (LAB) reduction96 to furnish secondary amine 68 in a straightforward deprotection procedure. Gratifyingly, access to the overall syn-diastereomer could also be obtained through inversion of the oxygen-bearing carbon stereocenter of 68 by boc-protection of the amine followed by anchimeric-assisted internal substitution of the hydroxyl group utilizing SOCl2 activation.97
 | ||
| Scheme 10 Improved enantiocontrol in the Cu-catalysed reductive coupling with ketones using an acyclic allenamide. | ||
The diastereodivergency enabled through the nature of the N-substituents of the allenamide (i.e. acyclic vs. cyclic) could be rationalized considering chair-like transition state models depicted as their Newman projections in Scheme 11.48 Stereocontrol in Cu-catalysed reductive allylation reactions is typically a result of Curtin–Hammett kinetics for the addition of rapidly equilibrating substituted allylic copper nucleophilic intermediates to the electrophile.45,67 As such, the anti/syn diastereocontrol is a function of the olefin geometry of the N-substituted allylic intermediates with the E-enamide leading to anti-products (e.g.(E)-AA-TS and (E)-CC-TS) and the Z-enamide leading to syn-products (e.g.(Z)-AA-TS and (Z)-CC-TS). The preference of the reaction involving acyclic allenamides for the anti-diastereomer seems counterintuitive considering that the transition state leading to this product has two gauche interactions (i.e.(E)-AA-TS) compared to the one gauche interaction present in the transition state furnishing the syn-diastereomer (i.e.(Z)-AA-TS). However, when considering that Z-enamides are known to exist as “twisted” amides to due to penalizing A1,3-strain,98,99 this twisting leads to steric shielding of the ketone approach vector by the R1-group present in acyclic allenamides to destabilize (Z)-AA-TS and cause the anti-diastereomer of product to be favored. In contrast, the analogous syn-forming transition structure for cyclic allenamides (i.e.(Z)-CC-TS) has reduced steric shielding of the ketone approach vector since the substituents are tied back in a ring and rotated away from the approaching ketone. This presumably allows for (Z)-CC-TS to be lower in energy to (E)-CC-TS that contains the larger number of gauche interactions affording overall syn-diastereoselectivity.
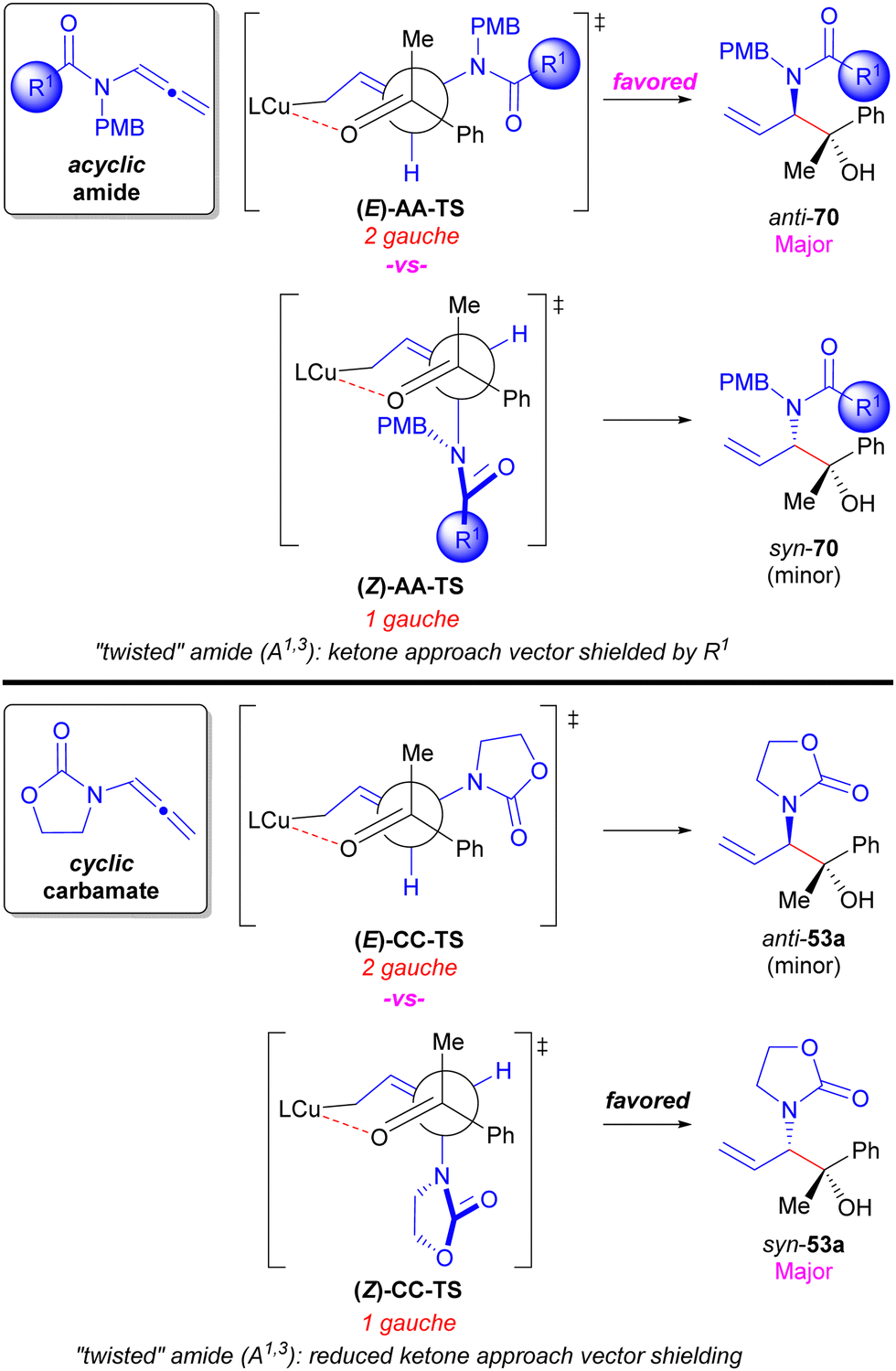 | ||
| Scheme 11 Diastereodivergency model for cyclic vs acyclic allenamides in Cu-catalysed asymmetric reductive coupling with ketone electrophiles. | ||
While Cu-catalysed reductive coupling reactions involving ketones produced aminoalcohols bearing tertiary alcohols, it is also desirable to expand the methodology towards generating those containing secondary alcohols by using aldehydes as coupling partners. Aldehydes are typically difficult coupling partners for CuH-catalysis due to competitive reduction of the aldehyde over hydrometalation of the unsaturated hydrocarbon coupling partner.64 Of the rare examples of use of aldehydes in CuH-catalysis,68,100 this can be circumvented by performing slow addition of the aldehyde to the reaction mixture. Utilizing this approach, a CuH-catalysed enantioselective reductive coupling of aldehydes and a N-Boc derived acyclic allenamide (71) was developed to provide access to chiral 1,2-amionalcohols bearing a secondary alcohol motif (72, Scheme 12).50 Again, the anti-diastereomer of product was the major diastereomer formed when using the acyclic allenamide 71. The yields and enantioselectivities of this process ranged from good to excellent and both aromatic and aliphatic aldehydes could be used. Furthermore, the N-Boc group could be easily removed using TFA to access the free secondary amine of 73. Notably, as with the previous acyclic allenamide/ketone coupling system, a syn-derivative (74) could also be accessed from the anti-products (e.g.72g) by anchimeric-assisted internal substitution of the secondary alcohol by the N-Boc group.97 This invertive process was exploited in the context of studies towards the synthesis of eligulstat (77), an important compound for the treatment of Gaucher's disease.101 Aminoalcohol precursor 72h was prepared in high yield and enantioselectivity using the established reductive coupling process, followed by anchimeric-assisted cyclocarbamation to provide 75 having the requisite relative stereochemistry required for eligulstat. Subsequent ozonolysis of 75 followed by a reductive amination workup102 with pyrrolidine afforded 76 as a potential precursor towards eligulstat.
 | ||
| Scheme 12 Enantioselective Cu-catalysed reductive coupling of aldehydes with an acyclic allenamide. | ||
The 1,2-aminoalcohol reaction products from aldehyde aminoallylation under Cu-catalysis (Scheme 12) have also been achieved utilizing Krische's hydrogen autotransfer18 process initially in a non-enantioselective fashion employing Ru-catalysis,54,70,71 and more recently in an enantioselective fashion3 using Ir-catalysis (Scheme 13). By employing the N-phthaloyl derived allenimide 78, chiral anti-1,2-aminoalcohol synthons can be accessed in excellent enantioselectivities.3 Here the aldehyde coupling partner is employed at the alcohol oxidation state which is oxidized in situ through dehydrogenation by the catalyst to generate the necessary metal-hydride intermediate needed to hydrometalate the allene and provide the allylmetal nucleophile for addition to the formed aldehyde. Such hydrogen-borrowing processes are highly atom economical and attractive tools for complex molecule synthesis.
 | ||
| Scheme 13 Krische's enantioselective Ir-catalysed aminoallylation of aldehydes using hydrogen autotransfer with an allenimide. | ||
Besides the use of allenamides for the introduction of N-heteroatoms using reductive coupling, the Malcolmson group recently introduced the novel 2-azadiene103,104 (80) and 2-azatriene105 (84) reagents for the asymmetric synthesis of 1,2-aminoalcohols and 1,2-diamines by Cu-catalysed enantioselective reductive coupling (Scheme 14). While enantioselective coupling reactions with 2-azadienes (80) has been achieved with both ketone103 and imine104 electrophiles, saturated 1,2-aminoalcohols (83) or 1,2-diamines (82) are obtained. In contrast, the use of 2-azatrienes (84) provides access to unsaturated products 85 in reactions with imine105 electrophiles (81). Notably, a diastereodivergent process could be developed employing 2-azatriene reagents 84 to afford either anti-85 or syn-85 by tuning the chiral ligand used for catalysis (L1vs.L2, respectively). Furthermore, 2-azatriene reagents offer the possibility to access regioisomeric products according to the strategy outlined in Fig. 3B as a tool to prepare multiple different heteroatom substitution patterns, however, regiodivergent asymmetric processes employing 84 to exploit this potential have not yet been reported.
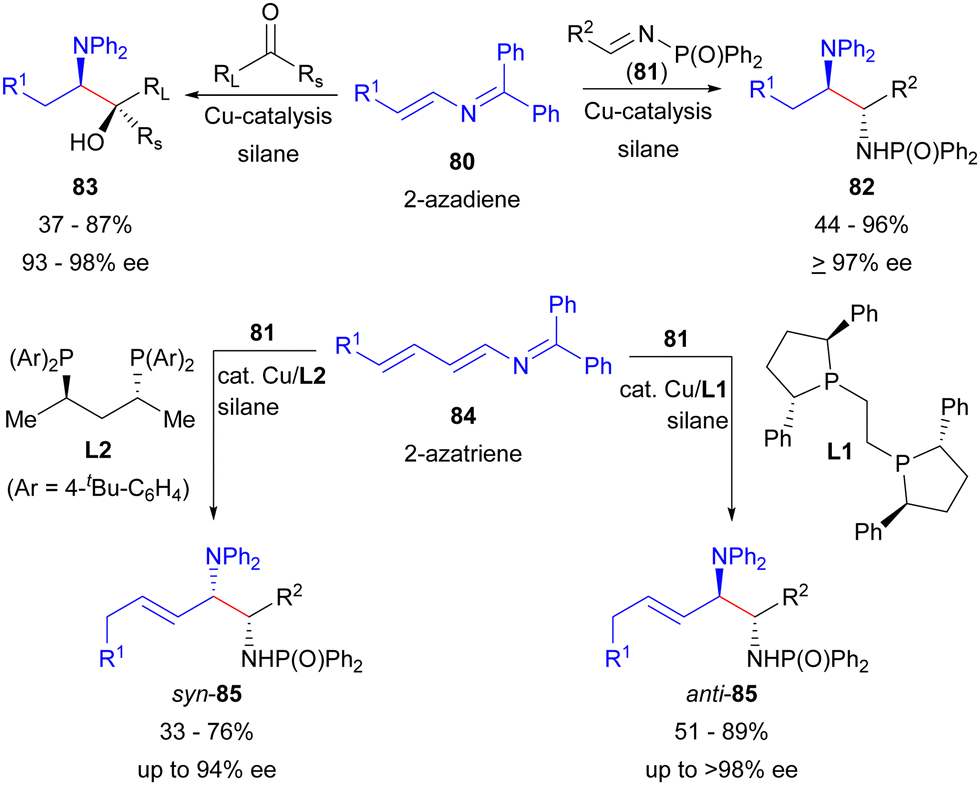 | ||
| Scheme 14 Malcolmson's enantioselective Cu-catalysed reductive coupling using 2-azadiene and 2-azatriene reagents. | ||
Recently, photoredox catalysis has been utilized as a technique to enable Cr-catalysed allylation reactions by hydrogen atom abstraction (HAT, Scheme 15).106,107 While this process is overall redox neutral, use of allylamine precursors (86) allows for branched-selective aldehyde aminoallylation in a non-enantioselective fashion.106 While an enantioselective variant was achieved with allylsilanes (Hosomi-Sakurai reaction),107 the enantioselective variant with a heteroatom-substituted allylic silane was not provided.
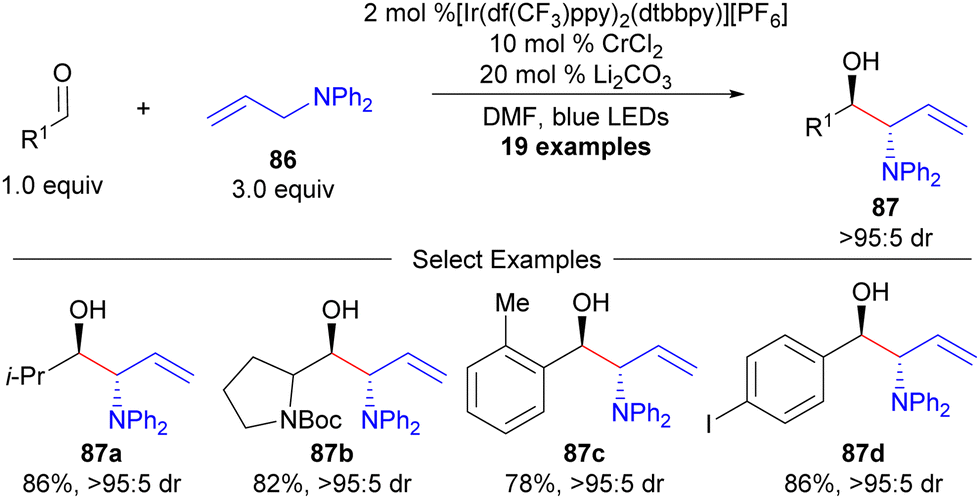 | ||
| Scheme 15 Glorius’ redox neutral aminoallylation by dual photoredox and chromium catalysis. | ||
Enantioselective reductive coupling of allenamides utilizing alternative reductants
All the previously discussed reductive coupling reactions employing N-substituted allenes utilized hydride based reducing agents to afford H-substituted products. However, reductive coupling can utilize alternative reductants to allow for the incorporation of additional functionality (Fig. 3A). For example, diboron reagents have been utilized in Type 1 reductive coupling processes108,109 leading to products containing C–B functional group(s).18 Accordingly, borylcupration110 processes are well established, and therefore, utilizing diboronate reducing agents in place of silane for Cu-catalysed reductive coupling reaction of N-substituted allenes (88) can provide two main advantages over the use of silane reductants: (1) aldehyde reduction may be circumvented, and (2) addition of a C–B motif into the final product (e.g.90) that offers significant opportunities for further functionalization due to the vast diversity of transformations the C–B bond can undergo (Scheme 16).49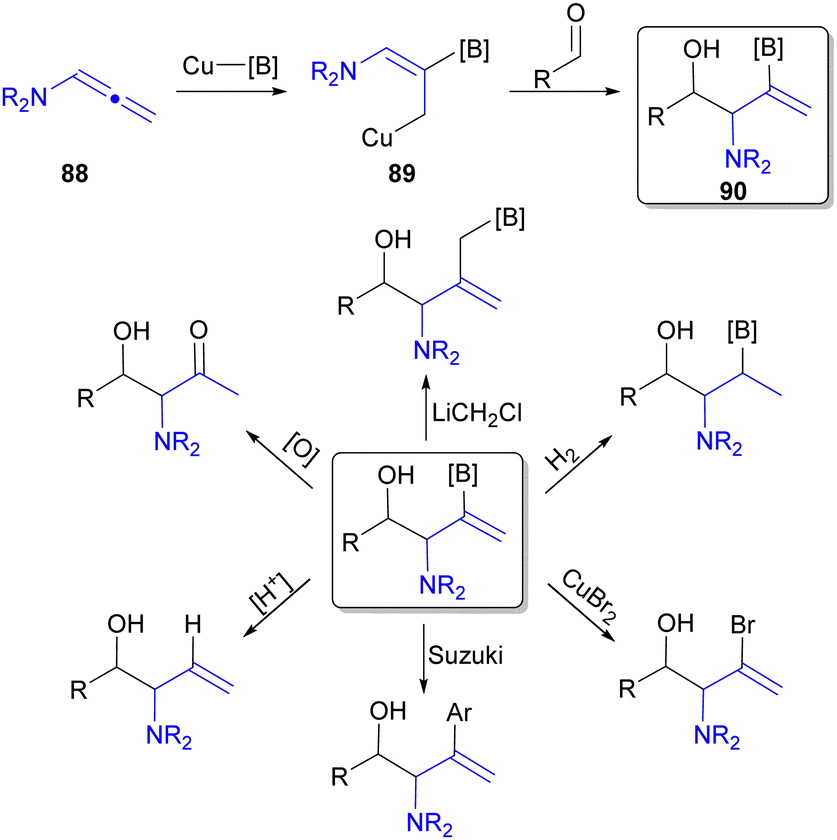 | ||
| Scheme 16 Synthetic opportunities available to Cu-catalysed reductive borylation of N-substituted allenes. | ||
While borylative Cu-catalysed enantioselective reductive coupling reactions of carbon-substituted allenes using both aldehyde or imine electrophiles was initially reported by the groups of Hoveyda111 and Procter,112 respectively, reactions employing N-substituted allenes have been less developed113,114 prior to our work.49 Enantioselective Cu-catalysed borylative reductive coupling of allenimide 78 with aldehyde electrophiles was identified to proceed in high stereoselectivities using Ph-BPE (L2) as ligand to provide intermediate 91 (Scheme 17). This intermediate was found to be quite sensitive to protonolysis during silica gel chromatography, and therefore, further functionalization reactions of the C–B moiety were developed using telescoped processes through direct transformation of crude 91. Protonolysis using AcOH led to products 92 from formal reductive coupling employing hydride reductants (e.g.79, Scheme 13). Oxidation of 91 using H2O2 led to formal aldol products 93 whereas direct Suzuki–Miyaura cross coupling of 91 with aryl iodides or bromides allowed access to 1,1-disubstituted alkene compounds 94.
 | ||
| Scheme 17 Cu-catalysed enantioselective reductive borylation of aldehydes with an N-phthaloyl allene. | ||
The sensitivity of 91 to undergo protonolysis was hypothesized to result from the neighboring free hydroxyl group present.115 Consequently, direct protection of the alcohol group as a TBS-ether through addition of TBSOTf to the completed reductive coupling reaction allowed for the isolation of intermediate 91 as the silyl ether (95). Additional transformations of the reaction products from the reductive borylation sequences were also demonstrated to enable access to heteroatom-rich compounds 96 and 97 from a B-aldol reaction or olefin hydroboration of ent-93c and ent-94d, respectively.
An additional interesting feature of the Cu-catalysed reductive borylation of allenimide 78 and aldehydes was the observed selective formation of the anti-diastereomer of product (91). Previous Cu-catalysed reductive borylation reactions of carbon-substituted allenes with aldehydes were demonstrated to afford the syn-diastereomer of product as the major isomer.111 This typically is rationalized through addition of a substituted allylic copper nucleophile (C-99) to the aldehyde through a chair-like transition structure (C-TS, Scheme 18). Preference for the Z-alkene geometry of nucleophile C-99 can be explained on steric grounds where the bulky (pin)B-moiety prefers a trans-relationship to the allene substituent (Rc).111 In contrast, the turnover in diastereoselectivity with N-phthaloyl derived allene 78 was rationalized through a catalyst directing effect by the N-phthaloyl group (N-98) to favour nucleophilic Cu-intermediate N-99 having E-alkene geometry that may also benefit from carbonyl lone-pair donation into the vacant boron p-orbital.49 Addition of N-99 to aldehyde through N-TS leads to the observed anti-stereocontrol in reactions employing allene 78. As a result, this demonstrates that not only can directing groups on the allene coupling partner be used to control regioselectivity (Fig. 3b, 4) but can also be used as a tool to impact stereocontrol.
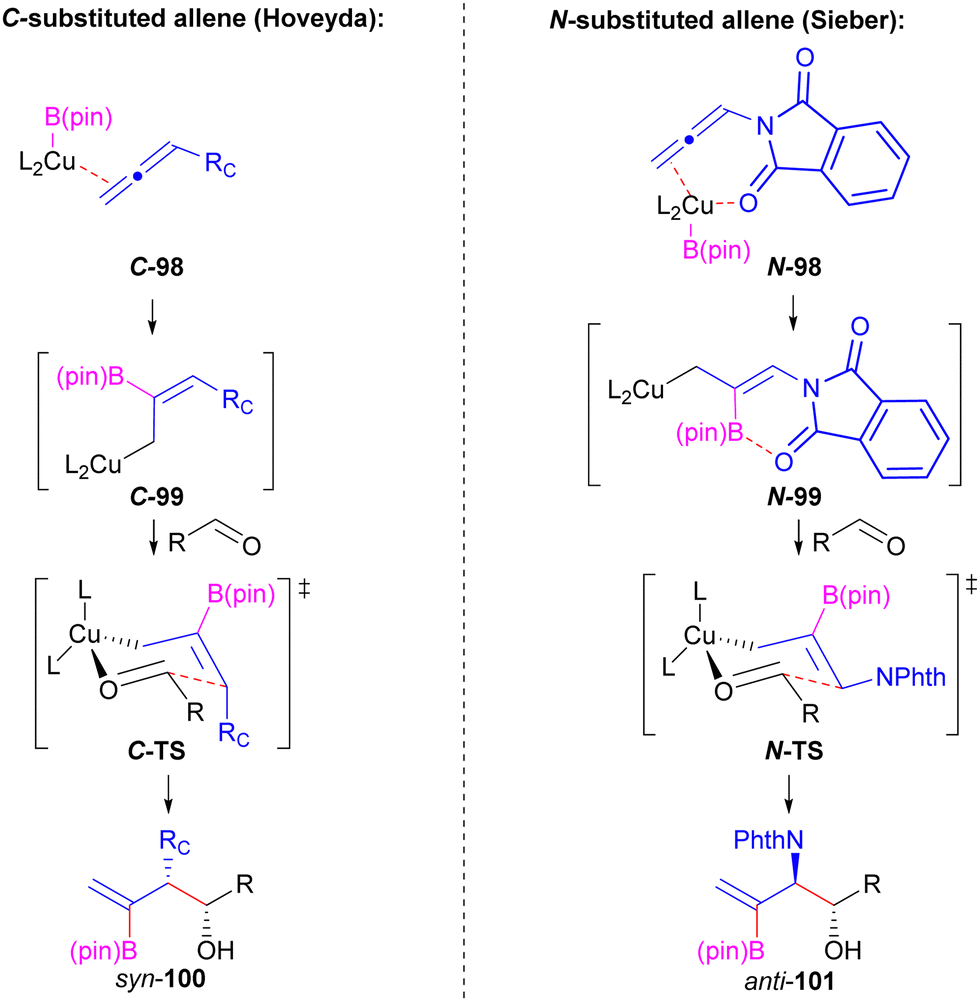 | ||
| Scheme 18 Diastereoselectivity differences between C- versus N-substituted allenes in Cu-catalysed reductive borylation. | ||
In an alternative process developed by Chen,113 access to similar products to 91 have also been obtained by a tandem Pt-catalyzed allene diboration/allylboration sequence (103, Scheme 19). While this pathway enables access to the syn-diastereomer of product, only racemates are obtained. Alternatively, Murakami114 developed a strategy utilizing allylic transposition of vinyl diboronate 104 to 106 that can then be trapped in situ with aldehyde electrophiles to access anti-products 107 in a racemic fashion (Scheme 19). The opposite relative stereocontrol relative to Chen's process is a result of selective formation of the opposite alkene geometry (106) relative to 102 provided by the Ru-catalyst in the allylic transposition. The starting diboron reagent 104 used was previously prepared using Pt-catalysed diboration of alkyne 105.116
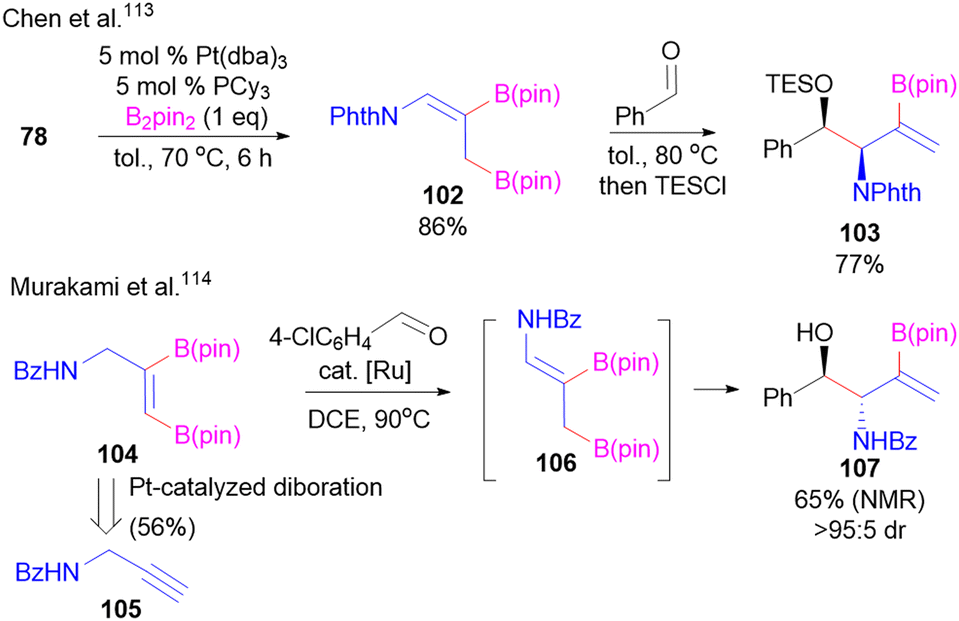 | ||
| Scheme 19 Strategy towards racemic boryl-substituted 1,2-aminoalcohols through Pt-catalysed diboration/aldehyde allylboration. | ||
Conclusions
In this feature article, we demonstrate that the use of N-substituted allenes in asymmetric reductive coupling reactions represent a convenient tool towards enabling a synthetic strategy whereby multiple heteroatom substitution patterns can be generated using the same reaction starting materials. Accordingly, the use of an appropriate N-substituent to serve as a directing group and by catalyst modification allows for tuning of regiodivergency in these processes and can also lead to differences in diastereoselection. Previous methods to access similar products typically required the use of strong bases and cryogenic reaction temperatures to generate preformed organometallic nucleophiles from aza-allyl anion equivalents. Modern reductive coupling processes allow for increased functional group compatibility and more convenient setup by operating under ambient conditions. Overall, the development of reductive coupling asymmetric processes of heteroatom-substituted unsaturated hydrocarbons are still relatively underdeveloped. For instance, only reactions of terminal allenamides have currently been reported, and investigation into alternative electrophile addition pathways, such as 1,4-conjugate addition when employing α,β-unsaturated carbonyl compounds have not yet been disclosed. While both 1,2- and 1,4-heteroatom substitution has been demonstrated in these processes, significantly more methods are available for generation of 1,2-substituted products over the 1,4-substituted derivatives. Future work in this area is needed to enable additional processes and strategies to increase the types of heteroatom substitution patterns that can be accessed (e.g. 1,3-; 1,4-; 1,5-; 1,6-substitution, etc.), and expand the scope of the heteroatom-substituted unsaturated coupling partners employed (e.g. 1,1-disubstituted or chiral 1,3-disubstituted allenamides).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank past and present group members for their significant contributions, as their names appear in cited references from our group. We acknowledge laboratory startup funding that has supported this work from the Virginia Commonwealth University and the Bill and Melinda Gates Foundation (Medicines for All Institute Grant, OPP#1176590). We also acknowledge the National Science Foundation (CHE-2143712) and REU supported students (CHE-1851916) for additional support that has contributed to this work. Solvias is acknowledged for the generous donation of W12.Notes and references
- E. K. Davison and J. Sperry, J. Nat. Prod., 2017, 80, 3060–3079 CrossRef CAS PubMed.
- S. R. S. Saibabu Kotti, C. Timmons and G. Li, Chem. Biol. Drug Des., 2006, 67, 101–114 CrossRef CAS PubMed.
- K. Spielmann, M. Xiang, L. A. Schwartz and M. J. Krische, J. Am. Chem. Soc., 2019, 141, 14136–14141 CrossRef CAS PubMed.
- J. B. Hendrickson, J. Am. Chem. Soc., 1975, 97, 5784–5800 CrossRef CAS.
- T. Gaich and P. S. Baran, J. Org. Chem., 2010, 75, 4657–4673 CrossRef CAS PubMed.
- R. Dach, J. J. Song, F. Roschangar, W. Samstag and C. H. Senanayake, Org. Process Res. Dev., 2012, 16, 1697–1706 CrossRef CAS.
- D. Urabe, T. Asaba and M. Inoue, Chem. Rev., 2015, 115, 9207–9231 CrossRef CAS PubMed.
- Y. Gao and D. Ma, Acc. Chem. Res., 2021, 54, 569–582 CrossRef CAS PubMed.
- D. Seebach, Angew. Chem., Int. Ed. Engl., 1979, 18, 239–258 CrossRef.
- D. A. Evans and G. C. Andrews, Acc. Chem. Res., 1974, 7, 147–155 CrossRef CAS.
- S. E. Dibrell, Y. Tao and S. E. Reisman, Acc. Chem. Res., 2021, 54, 1360–1373 CrossRef CAS PubMed.
- L. C. Campeau and N. Hazari, Organometallics, 2019, 38, 3–35 CrossRef CAS PubMed.
- N. Miyaura, K. Yamada and A. Suzuki, Tetrahedron Lett., 1979, 20, 3437–3440 CrossRef.
- E. Negishi, A. O. King and N. Okukado, J. Org. Chem., 1977, 42, 1821–1823 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 50, 4467–4470 CrossRef.
- A. Lei, W. Shi, C. Liu and W. Liu, Oxidative Cross-Coupling Reactions, Wiley-VCH, Verlag GmbH& Co., 2016 Search PubMed.
- I. Funes-Ardoiz and F. Maseras, ACS Catal., 2018, 8, 1161–1172 CrossRef CAS.
- T. Agrawal and J. D. Sieber, Synthesis, 2020, 2623–2638 CAS.
- M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026–6052 CrossRef CAS PubMed.
- Y. Sato, M. Takimoto, M. Mori, K. Hayashi, T. Katsuhara and K. Takagi, J. Am. Chem. Soc., 1994, 116, 9771–9772 CrossRef CAS.
- M. Kimura, A. Ezoe, K. Shibata and Y. Tamara, J. Am. Chem. Soc., 1998, 120, 4033–4034 CrossRef CAS.
- E. Oblinger and J. Montgomery, J. Am. Chem. Soc., 1997, 119, 9065–9066 CrossRef CAS.
- K. M. Miller, W. S. Huang and T. F. Jamison, J. Am. Chem. Soc., 2003, 125, 3442–3443 CrossRef CAS PubMed.
- N. M. Kablaoui and S. L. Buchwald, J. Am. Chem. Soc., 1995, 117, 6785–6786 CrossRef CAS.
- S. Kang and S. Yoon, Chem. Commun., 2002, 2634–2635 RSC.
- S. Ogoshi, M. A. Oka and H. Kurosawa, J. Am. Chem. Soc., 2004, 126, 11802–11803 CrossRef CAS PubMed.
- J. Montgomery, Angew. Chem., Int. Ed., 2004, 43, 3890–3908 CrossRef CAS PubMed.
- A. Krasovskiy, C. Duplais and B. H. Lipshutz, J. Am. Chem. Soc., 2009, 131, 15592–15593 CrossRef CAS PubMed.
- W. M. Czaplik, M. Mayer and A. J. Von Wangelin, Angew. Chem., Int. Ed., 2009, 48, 607–610 CrossRef CAS PubMed.
- W. M. Czaplik, M. Mayer, S. Grupe and A. J. Von Wangelin, Pure Appl. Chem., 2010, 82, 1545–1553 CAS.
- X. Yu, T. Yang, S. Wang, H. Xu and H. Gong, Org. Lett., 2011, 13, 2138–2141 CrossRef CAS PubMed.
- D. A. Everson, R. Shrestha and D. J. Weix, J. Am. Chem. Soc., 2010, 132, 920–921 CrossRef CAS PubMed.
- D. A. Everson and D. J. Weix, J. Org. Chem., 2014, 79, 4793–4798 CrossRef CAS PubMed.
- C. E. I. Knappke, S. Grupe, D. Gärtner, M. Corpet, C. Gosmini and A. Jacobi Von Wangelin, Chem. – Eur. J., 2014, 20, 6828–6842 CrossRef CAS PubMed.
- X. Wang, Y. Dai and H. Gong, Top. Curr. Chem., 2016, 374, 1–29 CrossRef CAS PubMed.
- E. Richmond and J. Moran, Synthesis, 2018, 499–513 CrossRef CAS.
- R. T. Smith, X. Zhang, J. A. Rincón, J. Agejas, C. Mateos, M. Barberis, S. García-Cerrada, O. De Frutos and D. W. C. Macmillan, J. Am. Chem. Soc., 2018, 140, 17433–17438 CrossRef CAS PubMed.
- H. A. Sakai, W. Liu, C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2020, 142, 11691–11697 CrossRef CAS PubMed.
- A. Jutand, Chem. Rev., 2008, 108, 2300–2347 CrossRef CAS PubMed.
- R. J. Perkins, D. J. Pedro and E. C. Hansen, Org. Lett., 2017, 19, 3755–3758 CrossRef CAS PubMed.
- R. J. Perkins, A. J. Hughes, D. J. Weix and E. C. Hansen, Org. Process Res. Dev., 2019, 23, 1746–1751 CrossRef CAS.
- T. J. Delano and S. E. Reisman, ACS Catal., 2019, 9, 6751–6754 CrossRef CAS PubMed.
- R. K. Klake, S. L. Gargaro, S. L. Gentry, S. O. Elele and J. D. Sieber, Org. Lett., 2019, 21, 7992–7998 CrossRef CAS PubMed.
- S. L. Gargaro, R. K. Klake, K. L. Burns, S. O. Elele, S. L. Gentry and J. D. Sieber, Org. Lett., 2019, 21, 9753–9758 CrossRef CAS PubMed.
- T. Agrawal, R. T. Martin, S. Collins, Z. Wilhelm, M. D. Edwards, O. Gutierrez and J. D. Sieber, J. Org. Chem., 2021, 86, 5026–5046 CrossRef CAS PubMed.
- R. K. Klake, M. D. Edwards and J. D. Sieber, Org. Lett., 2021, 23, 6444–6449 CrossRef CAS PubMed.
- D. B. Ho, S. Gargaro, R. K. Klake and J. D. Sieber, J. Org. Chem., 2022, 87, 2142–2153 CrossRef CAS PubMed.
- S. Collins and J. D. Sieber, Org. Lett., 2023, 25, 1425–1430 CrossRef CAS PubMed.
- S. L. Gargaro, R. K. Klake and J. D. Sieber, Org. Lett., 2023, 25, 4644–4649 CrossRef CAS PubMed.
- R. K. Klake and J. D. Sieber, Org. Lett., 2023, 25, 4730–4734 CrossRef CAS PubMed.
- T. Lu, Z. Lu, Z.-X. Ma, Y. Zhang and R. P. Hsung, Chem. Rev., 2013, 113, 4862–4904 CrossRef CAS PubMed.
- B. Alcaide and P. Almendros, Adv. Synth. Catal., 2011, 353, 2561–2576 CrossRef CAS.
- R. Blieck, M. Taillefer and F. Monnier, Chem. Rev., 2020, 120, 13545–13598 CrossRef CAS PubMed.
- E. Skucas, J. R. Zbieg and M. J. Krische, J. Am. Chem. Soc., 2009, 5054–5055 CrossRef CAS PubMed.
- L. A. Schwartz, M. Holmes, G. A. Brito, T. P. Gonçalves, J. Richardson, J. C. Ruble, K. W. Huang and M. J. Krische, J. Am. Chem. Soc., 2019, 141, 2087–2096 CrossRef CAS PubMed.
- S. W. Kim, C. C. Meyer, B. K. Mai, P. Liu and M. J. Krische, ACS Catal., 2019, 9, 9158–9163 CrossRef CAS PubMed.
- M. Xiang, D. E. Pfaffinger, E. Ortiz, G. A. Brito and M. J. Krische, J. Am. Chem. Soc., 2021, 143, 8849–8854 CrossRef CAS PubMed.
- J. R. Zbieg, J. Moran and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 10582–10586 CrossRef CAS PubMed.
- E. L. McInturff, E. Yamaguchi and M. J. Krische, J. Am. Chem. Soc., 2012, 134, 20628–20631 CrossRef CAS PubMed.
- K. D. Nguyen, D. Herkommer and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 14210–14213 CrossRef CAS PubMed.
- L. M. Geary, S. K. Woo, J. C. Leung and M. J. Krische, Angew. Chem., Int. Ed., 2012, 51, 2972–2976 CrossRef CAS PubMed.
- L. M. Geary, J. C. Leung and M. J. Krische, Chem. – Eur. J., 2012, 18, 16823–16827 CrossRef CAS PubMed.
- K. D. Nguyen, D. Herkommer and M. J. Krische, J. Am. Chem. Soc., 2016, 138, 5238–5241 CrossRef CAS PubMed.
- Y. Yang, I. B. Perry, G. Lu, P. Liu and S. L. Buchwald, Science, 2016, 353, 144–150 CrossRef CAS PubMed.
- E. Y. Tsai, R. Y. Liu, Y. Yang and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 2007–2011 CrossRef CAS PubMed.
- R. Y. Liu, Y. Zhou, Y. Yang and S. L. Buchwald, J. Am. Chem. Soc., 2019, 141, 2251–2256 CrossRef CAS PubMed.
- C. Li, R. Y. Liu, L. T. Jesikiewicz, Y. Yang, P. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2019, 141, 5062–5070 CrossRef CAS PubMed.
- C. Li, K. Shin, R. Y. Liu and S. L. Buchwald, Angew. Chem., Int. Ed., 2019, 58, 17074–17080 CrossRef CAS PubMed.
- H. Iwamoto, Y. Hayashi, Y. Ozawa and H. Ito, ACS Catal., 2020, 10, 2471–2476 CrossRef CAS.
- J. R. Zbieg, E. L. Mcinturff and M. J. Krische, Org. Lett., 2010, 12, 2514–2516 CrossRef CAS PubMed.
- W. Zhang, W. Chen, H. Xiao and M. J. Krische, Org. Lett., 2017, 19, 4876–4879 CrossRef CAS PubMed.
- M. Nerz-stormes and E. R. Thornton, J. Org. Chem., 1991, 56, 2489–2498 CrossRef CAS.
- C. Gaul and D. Seebach, Helv. Chim. Acta, 2002, 85, 963–978 CrossRef CAS.
- R. Dorta, N. M. Scott, C. Costabile, L. Cavallo, C. D. Hoff and S. P. Nolan, J. Am. Chem. Soc., 2005, 127, 2485–2495 CrossRef CAS PubMed.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- D. A. Evans, J. M. Takacs, L. R. Mcgee, M. D. Ennis, D. J. Mathre and J. Bartroli, Pure Appl. Chem., 1981, 53, 1109–1127 CrossRef CAS.
- D. Hoppe, Angew. Chem., Int. Ed. Engl., 1984, 23, 932–948 CrossRef.
- J. Y. Kang and B. T. Connell, J. Am. Chem. Soc., 2010, 132, 7826–7827 CrossRef CAS PubMed.
- D. Hoppe and O. Zschage, Angew. Chem., Int. Ed. Engl., 1989, 28, 69–71 CrossRef.
- E. D. Burke, N. K. Lim and J. L. Gleason, Synlett, 2003, 390–392 CAS.
- G. Piancatelli, A. Scettri and M. D’Auria, Tetrahedron Lett., 1977, 18, 3483–3484 CrossRef.
- D. A. Mulholland, K. McFarland and M. Randrianarivelojosia, Biochem. Syst. Ecol., 2006, 34, 365–369 CrossRef CAS.
- A. G. M. Barrett, M. A. Seefeld and D. J. Williams, J. Chem. Soc., Chem. Commun., 1994, 1053–1054 RSC.
- A. G. M. Barrett, M. A. Seefeld, A. J. P. White and D. J. Williams, J. Org. Chem., 1996, 61, 2677–2685 CrossRef CAS PubMed.
- G. A. Weisenburger and P. Beak, J. Am. Chem. Soc., 1996, 118, 12218–12219 CrossRef CAS.
- D. J. Pippel, G. A. Weisenburger, N. C. Faibish and P. Beak, J. Am. Chem. Soc., 2001, 123, 4919–4927 CrossRef CAS PubMed.
- E. Ascic and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 4666–4669 CrossRef CAS PubMed.
- R. Y. Liu, Y. Yang and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 14077–14080 CrossRef CAS PubMed.
- T. L. Cottrell, The Strength of Chemical Bonds, 2nd edn, 1958 Search PubMed.
- M. C. Desai, S. L. Lefkowitz, P. F. Thadeio, K. P. Longo and R. M. Snider, J. Med. Chem., 1992, 35, 4911–4913 CrossRef CAS PubMed.
- M. B. Uphade, A. A. Reddy, S. P. Khandare and K. R. Prasad, Org. Lett., 2019, 21, 9109–9113 CrossRef CAS PubMed.
- M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600–3740 CrossRef CAS PubMed.
- R. M. Beesley, C. K. Ingold and J. F. Thorpe, J. Chem. Soc., Trans., 1915, 107, 1080–1106 RSC.
- B. L. Shaw, J. Am. Chem. Soc., 1975, 97, 3856–3857 CrossRef CAS.
- K. Nogi and H. Yorimitsu, Chem. Rev., 2021, 121, 345–364 CrossRef CAS PubMed.
- A. G. Myers, B. H. Yang and D. J. Kopecky, Tetrahedron Lett., 1996, 37, 3623–3626 CrossRef CAS.
- S. Kano, Y. Yuasa, T. Yokomatsu and S. Shibuya, Heterocycles, 1988, 27, 1241–1248 CrossRef CAS.
- Z. Song, T. Lu, R. P. Hsung, Z. F. Al-Rashid, C. Ko and Y. Tang, Angew. Chem., 2007, 119, 4147–4150 CrossRef.
- A. J. Clark, D. P. Curran, D. J. Fox, F. Ghelfi, C. S. Guy, B. Hay, N. James, J. M. Phillips, F. Roncaglia, P. B. Sellars, P. Wilson and H. Zhang, J. Org. Chem., 2016, 81, 5547–5565 CrossRef CAS PubMed.
- W. J. Jang and J. Yun, Angew. Chem., Int. Ed., 2018, 57, 12116–12120 CrossRef CAS PubMed.
- L. J. Scott, Drugs, 2015, 75, 1669–1678 CrossRef CAS PubMed.
- H. Yung-Son and L. Ling, Tetrahedron Lett., 1993, 34, 5309–5312 CrossRef.
- K. Li, X. Shao, L. Tseng and S. J. Malcolmson, J. Am. Chem. Soc., 2018, 140, 598–601 CrossRef CAS PubMed.
- X. Shao, K. Li and S. J. Malcolmson, J. Am. Chem. Soc., 2018, 140, 7083–7087 CrossRef CAS PubMed.
- P. Zhou, X. Shao and S. J. Malcolmson, J. Am. Chem. Soc., 2021, 143, 13999–14008 CrossRef CAS PubMed.
- J. L. Schwarz, F. Schäfers, A. Tlahuext-Aca, L. Lückemeier and F. Glorius, J. Am. Chem. Soc., 2018, 140, 12705–12709 CrossRef CAS PubMed.
- F. Schäfers, S. Dutta, R. Kleinmans, C. Mück-Lichtenfeld and F. Glorius, ACS Catal., 2022, 12, 12281–12290 CrossRef.
- Y. C. Hee and J. P. Morken, J. Am. Chem. Soc., 2008, 130, 16140–16141 CrossRef PubMed.
- H. Y. Cho, Z. Yu and J. P. Morken, Org. Lett., 2011, 13, 5267–5269 CrossRef CAS PubMed.
- D. Hemming, R. Fritzemeier, S. A. Westcott, W. L. Santos and P. G. Steel, Chem. Soc. Rev., 2018, 47, 7477–7494 RSC.
- F. Meng, H. Jang, B. Jung and A. H. Hoveyda, Angew. Chem., Int. Ed., 2013, 52, 5046–5051 CrossRef CAS PubMed.
- K. Yeung, R. E. Ruscoe, J. Rae, A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 11912–11916 CrossRef CAS PubMed.
- J. Chen, S. Gao, J. D. Gorden and M. Chen, Org. Lett., 2019, 21, 4638–4641 CrossRef CAS PubMed.
- T. Miura, J. Nakahashi, T. Sasatsu and M. Murakami, Angew. Chem., Int. Ed., 2019, 58, 1138–1142 CrossRef CAS PubMed.
- M. J. Hesse, S. Essafi, C. G. Watson, J. N. Harvey, D. Hirst, C. L. Willis and V. K. Aggarwal, Angew. Chem., Int. Ed., 2014, 53, 6145–6149 CrossRef CAS PubMed.
- B. M. Trost, J. J. Cregg and N. Quach, J. Am. Chem. Soc., 2017, 139, 5133–5139 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |