
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVitamin B12 and a metal–organic framework enable the photocatalytic generation of alkyl radicals†
Krzysztof R.
Dworakowski
a,
Artur
Chołuj
b,
Michał J.
Chmielewski
 *b and
Dorota
Gryko
*a
*b and
Dorota
Gryko
*a
aInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, Warsaw 01-224, Poland. E-mail: Dorota.gryko@icho.edu.pl
bFaculty of Chemistry, Biological and Chemical Research Centre, University of Warsaw, Żwirki i Wigury 101, Warsaw 02-089, Poland. E-mail: mchmielewski@chem.uw.edu.pl
First published on 22nd August 2023
Abstract
A versatile Co-catalyst-vitamin B12 (cobalamin)-can be photochemically reduced to its catalytically active Co(I) form under visible light irradiation, in the presence of MIL-125-NH2(Ti) as a photocatalyst and utilized for the generation of alkyl radicals. The prior reduction of cobalamin to the Co(II) form is not required in this method.
Vitamin B12 (1, cobalamin, Fig. 1) is a natural cobalt complex that acts as a cofactor in many biochemical processes including isomerisations, methyl transfer, and dehalogenation.1 Inspired by these natural processes, chemists have utilised this unique biomolecule as a Co-catalyst in multiple transformations,2,3 including dehalogenations,4 alkylations, dimerizations, strained ring openings,5,6 cyclopropanations,7,8 ester and amide formation,9 tandem addition to double bonds,10etc.11 The redox chemistry of the central cobalt cation plays a crucial role in these processes, and the oxidation state of the central metal ion defines the philicity and, consequently, the chemical behaviour of the entire molecule. Vitamin B12 in the +3 oxidation state can be reduced to either the radical Co(II) or supernucleophilic Co(I) species.1 The effective reduction of cobalamin(III) to the catalytically active Co(I) form can be achieved by either chemical, photochemical, or electrochemical means (Ep = −0.04 V vs. SCE for Co(III) to Co(II) and Ep = −0.85 V vs. SCE for Co(II) to Co(I).12 Thus, reducing agents, such as activated metals (Zn, Mn) or sodium borohydride, are most commonly used. However, some functional groups, such as aldehydes, halides, and disulphides, are not compatible with the reductive conditions.
 | ||
| Fig. 1 (A) Vitamin B12 (1, cyanocobalamin); (B) MIL-125-NH2(Ti) structure, grey-carbon, white-hydrogen, red-oxygen, light blue octahedral-titanium, blue-nitrogen. | ||
Recently, photochemical approaches to the generation of the supernucleophilic Co(I) species have been broadly investigated. The Hisaeda group reported the photochemical reduction of vitamin B12 derivatives under (a) homogeneous conditions, utilizing iridium13 or ruthenium9 photocatalysts and blue light irradiation, and (b) heterogeneous conditions using TiO2 and ultraviolet (UV) irradiation.14 These approaches, however, usually require: (1) pre-reduction of the Co(III) to the Co(II) state, or (2) irradiation with a highly energetic UV light, or (3) the use of precious transition metals as photocatalysts. Recently, Barata–Vallejo et al. described the first photochemical reduction of vitamin B12, which was achieved via electron transfer from Rose Bengal in the excited state, with TMEDA as a sacrificial reductant under green light irradiation.15 As important as this advance is, the system appears to function only in water and is limited to perfluoroalkyl partners in aromatic substitution. Thus, a more general and greener photocatalytic system is needed for the vitamin B12 reduction, that will broaden the applications of this sustainable Co-catalyst.
Photoredox-active metal–organic frameworks (MOFs) have emerged as versatile photocatalysts due to their modular, easily tuneable architectures, ultrahigh porosity, good e–h separation, and visible light absorption capacity.16 This prompted us to explore vitamin B12-mediated catalysis with MOFs acting as electron transfer agents under visible light irradiation. Thus far, the only precedent of a photocatalytic system composed of vitamin B12 and a MOF, was published by Xu et al., who immobilised [Ru(bpy)3]+2 and heptamethyl cobyrinate perchlorate, a hydrophobic vitamin B12 derivative, on a mix-metal zirconium–ruthenium MOF (Zr4Ru2(bpdc)4 ·4C2NH8·9DMF, bpdc = biphenyl-4,4′-dicarboxylic acid).17 In this system, however, the MOF acts as a solid support only and the pre-prepared Co(II) complex is reduced to Co(I) by electron transfer from the Ru-photocatalyst.
Herein, we demonstrate the visible light-induced photochemical reduction of vitamin B12 employing MIL-125-NH2(Ti) as the sole photoredox catalyst and its use in the generation of alkyl radicals.
Among the numerous photocatalytic MOFs, Ti(IV)-based MIL-125-NH2 (MIL-125-NH2(Ti)) has attracted our attention because of its exceptional hydrolytic stability and the ability to absorb visible light due to the presence of amino groups in the organic linker (Fig. 1B). It has a band gap of 2.7 eV and its UV-Vis adsorption band extends to around 550 nm, exhibiting two maxima at 330 and 375 nm.18 Upon light irradiation, MIL-125-NH2(Ti) undergoes reversible ligand-to-metal charge transfer (LMCT) with transient formation of mixed-valent Ti+4/Ti+3 clusters, that makes it an attractive candidate for an electron transfer reagent.19 There are, indeed, several reports describing photocatalytic applications of this MOF, mainly focussing on CO2 reduction,20–22 N2 fixation,23 water remediation,24–26 hydrogen evolution,27 and the synthesis of N-benzyl-1-phenylmethane-imines.28,29
These reports on MIL-125-NH2(Ti) acting as a photoredox catalyst under blue light irradiation made us question whether it can serve as a photoreductant for cobalamin (1). To test this hypothesis, a mixture of vitamin B12 (1) and a large excess of MIL-125-NH2(Ti) in EtOH was irradiated with blue light (450 nm) for 15 minutes. The mixture turned orange-brown, suggesting efficient reduction of cobalamin(III) to cobalamin(II) (Fig. 2A, blue line). Comparison of the UV-Vis spectrum of the supernatant (with maximum at 470 nm) with the reported spectroscopic features of different forms of vitamin B12 (1) corroborates the generation of cobalamin in the +2 oxidation state.30 Thus, MIL-125-NH2(Ti) alone can reduce vitamin B12 (1), but only in a single electron transfer process. As the addition of amines have an impact on the catalytic property of MOFs,31 the reduction was performed in the presence of DIPEA. Efficient photogeneration of the Co(I) species occurred, as confirmed by the optical absorption of the reaction mixture at 390 nm (green line). To shed light on the role of DIPEA, a mixture of MIL-125-NH2(Ti) and DIPEA in EtOH was irradiated with blue light for 15 minutes. After this time, the MOF changed colour from yellow to blue – a characteristic for Ti(III) complexes (for details see ESI†).20 Charge transfer from DIPEA, acting as a sacrificial electron donor, fills electron holes on the MOF linkers and stabilizes the photogenerated Ti(III). The photoreduced Ti4+/Ti3+ MOF (blue) was separated, washed to remove DIPEA, and reacted with cob(II)alamin in the dark. The UV-vis spectrum of the supernatant shows that under these conditions, the MOF alone is able to reduce cob(II)alamin to its catalytically active Co(I) form, although the reduction is not complete (for details, see ESI†). Apparently, Ti3+ in the pre-reduced blue MOF, having a negatively charged framework, is a stronger reductant than the short-lived, charge separated Ti3+ species, which are formed in the absence of DIPEA. Despite that an excess of MOF was used, the reduction of the Co(II) species was not complete. We presume that the whole process takes place on the surface of the MOF, that and electron hopping through the porous semiconducting MOF is negligible. Reek, van der Vlugt, and Gascon have shown that even cobaloximes that are smaller than vitamin B12 do not penetrate the MIL-125-NH2(Ti) framework (6 Å).32
 | ||
| Fig. 2 (A) Measured UV-Vis spectra of cobalamin (1) with cobalt in different oxidation states; cob(III)alamin (orange), cob(II)alamin (blue), and cob(I)alamin (green). (B) A mixture of MIL-125-NH2(Ti), DIPEA in EtOH before and after irradiation with 450 nm LED irradiation for 15 min. | ||
Next, the newly developed MOF-based catalytic system for the photoreduction of vitamin B12 to the Co(I) intermediate was tested in a model C–C bond forming transformation. We chose a simple cyclization of N-substituted tosylamides (Table 1),33 as this intramolecular reaction was proved to be catalysed by cob(I)alamin. Indeed, in the presence of vitamin B12, MIL-125-NH2(Ti) and DIPEA, desired product 3a formed in 59% yield (Table 1, entry 1). Background experiments revealed that the reaction does not proceed without catalysts, sacrificial electron donor, Ar atmosphere, or light (entry 2). The reaction in the presence of TiO2 and 2-amino terephthalic acid gave only traces of product, pointing to the key role of the intimate connection between the organic ligands and titanium-oxide clusters (entry 3, TiO2 (−0.25 V vs. NHE), MIL-125-NH2 (−0.75 V vs. NHE)). Swapping MIL-125-NH2 for other titanium compound like [Ti(Cp)2]Cl2 or TiO2 while under proper irradiation, (entries 4 and 5) gave the same results. Evaluation of various reaction's parameters, such as Co-catalyst type, sacrificial electron donors, additives, reaction time, wavelength of light, and intensity of light (for full data, see ESI†) enabled the synthesis of the desired product in 90% yield. The use of simplified models of vitamin B12, such as cobaloxime or cobalt porphyrin, results in strongly decreased yields (entries 4 and 5). The presence of NH4Cl improves the yield (entry 8), yet other proton donors do not exhibit a similar behaviour (entries 9 and 10), a feature often encountered in Zn-assisted reductions of vitamin B12.5,10 Interestingly, among several other ammonium salts tested (see ESI†), only those with the NH4+ cation increased the yield, suggesting that the salt acts as a proton donor. For a closer investigation, reactions were performed in C2D5OD and in the presence of ND4Cl. We not only observed a strong kinetic isotope effect (>8 h reaction time vs. 45 min) but also 95% incorporation of the deuterium atom in the product (according to 1H NMR, see ESI†) corroborating the formation of a carbanion during the catalytic cycle. Other MOFs (MIL-101-NH2, UiO-66-NH2, MUV-11, MIL-101-SO3H/Na, HKUST-1) absorbing in the visible region do not catalyse the cyclization reaction (for details see ESI†), suggesting that the synergistic action of Ti clusters and 2-aminoterephthalate antennas is indispensable for efficient photoreduction of cobalamin.
|
|
||
|---|---|---|
| Entry | Deviation from optimal conditions | Yield (%) |
| Reaction conditions: B12 (1, 0.5 mol%), MIL-125-NH2(Ti) (2.5 mol%), substrate (2a, 0.25 mmol), DIPEA (2 equiv.), NH4Cl (0.5 equiv.), EtOH (2 ml), inert gas (Ar), 450 nm LED 100%, 45 min; yields determined by GC.a Isolated yield. | ||
| 1 | iPrOH, NH4Cl (53 mg, 4 equiv.), DIPEA (175 μl, 4 equiv.) 7 W single LED, 18 h | 59a |
| 2 | no 1 or no MIL or no DIPEA or no Ar or no light | 0 |
| 3 | TiO2 and amino terephthalic acid instead of MIL | Traces |
| 4 | [Ti(Cp)2]Cl2 instead of MIL, 450 nm and 525 nm | Traces |
| 5 | TiO2 instead of MIL-125-NH2, 254 nm irradiation | Traces |
| 6 | TPP(Co) instead of 1 | 20 |
| 7 | Cobaloxime ClCo(dmgH2)py instead of 1 | 3 |
| 8 | No NH4Cl | 49 |
| 9 | TEA instead of DIPEA | 49 |
| 10 | 4-cyanophenol instead of NH4Cl | 39 |
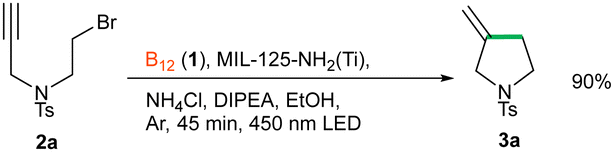
To confirm the crucial role of MIL-125-NH2(Ti) and the heterogeneous nature of the process, a split test was performed. Thus, the reaction mixture was irradiated for 25 min and then divided into two halves (yield 8%). One half of the mixture was irradiated for another 20 min, while the other half was filtrated before irradiation for another 20 min. The first part afforded product 3a in 87% yield, while the second part in only 10%, meaning that the reaction does not proceed without the solid MOF photocatalyst, and is not catalysed by any species leaking from the MOF during the reaction.
One of the most important features of the newly developed heterogeneous system is the possibility to recover the MOF catalyst. Thus, a set of three consecutive reactions was performed that gave the desired product in 69% (I run), is 74% (II run), and 83% (III run). Although the MOF was thoroughly washed after each round, the increase in yield might be attributed to the adsorption of vitamin B12 on the MOF surface, thus increasing the catalyst loading.
Based on the literature data and experimental indications, we propose the following mechanism for the cyclisation reaction (Scheme 1). First, the electron pair on the MOF linker is excited with 450 nm LED light, and LMCT from the amino group to the Ti(IV) node occurs. Next, DIPEA donates an electron and fills the positively charged hole on the linker. The Ti3+/Ti4+ nodes of the now negatively charged framework reduce vitamin B121 to cob(I)alamin IV. The latter reacts in an SN2 manner with alkyl bromide 2a to form Co–C complex V. The weak Co–C bond is then homolytically cleaved with 450 nm LED light to yield alkyl radical VI, which undergoes intramolecular cyclization followed by reduction and protonation to form final product 3a. The radical character of the reaction was corroborated by the radical trap experiment (see ESI†).
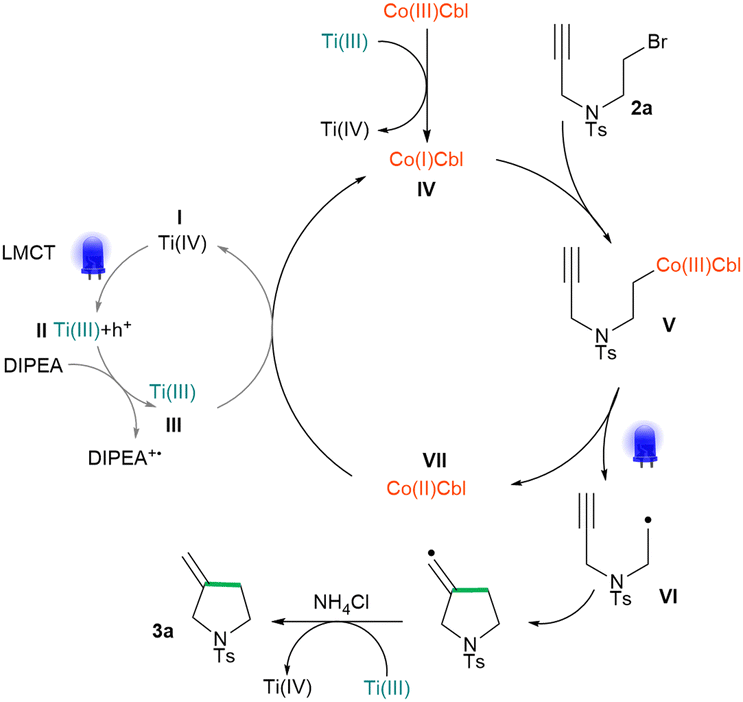 | ||
| Scheme 1 The proposed mechanism for radical cyclization. | ||
In the next step, several analogues of the model substrate were tested under the same reaction conditions (Table 2). In all six cases, very good to excellent yields were obtained. Interestingly, for compound 2c, the intermediate radical does not rearrange to the more stable benzyl radical. Substrate 2d yielded a side product with fully saturated cyclohexyl ring (3db), which is consistent with the literature precedents wherein olefinic precursor transforms into a mixture of saturated and unsaturated compounds. About group tolerance, aldehydes and chlorides are well tolerated while E-olefin isomerizes into Z-form (50%) and disulphide fully decomposes (for details see ESI†).
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yield (%) |
| Reaction conditions: B12 (1, 0.5 mol%), MIL-125-NH2(Ti) (6.0 μmol per molar weight), substrate (2a, 0.25 mmol), DIPEA (0.5 mmol, 2 equiv.), NH4Cl (0.125 mmol, 0.5 equiv.), EtOH (2 ml), inert gas (Ar), 450 nm LED, 2 h; isolated yields. | |||
| 1 |

|

|
85 |
| 2 |
|

|
85 E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) : :![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Z ∼ 1:1 Z ∼ 1:1 |
| 3 |

|

|
77 |
| 4 |

|
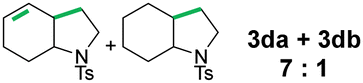
|
90 |
| 5 |

|
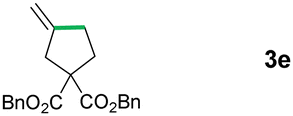
|
76 |
| 6 |

|

|
20 + 17 |

The excellent activity and heterogeneous nature of the MOF predisposed them to water and soil remediation for the degradation of organic pollutants. This is also true for cobalamin and its derivatives, which are effective in dechlorination reactions.34 Consequently, we used our newly developed system in the dehalogenation of 4,4′-dichlorodiphenyl-trichloroethane (5, 4,4′-DDT). Usually, the reaction leads either to a mixture of various derivatives4,35 or to dichlorodiphenyl-dichloroethane (4,4′-DDD) as the main product.13,17,36 In the presence of our newly developed catalytic system, the reaction yields three main products 6–8 with full substrate conversion (Scheme 2). Compound 8 has a dimeric structure and possesses only four chlorine atoms. Interestingly, mono dehalogenation (4,4′-DDD) was not observed, in contrast to previously known methods. This compound forms solely when the reaction was performed in the presence of only MOF (40% conversion), corroborating the crucial role of vitamin B12.
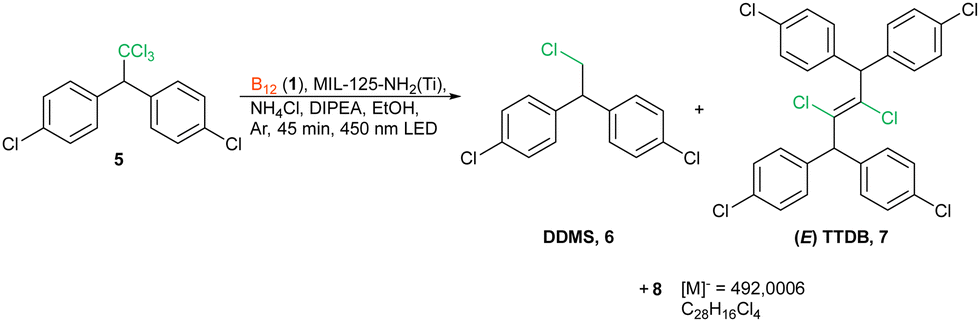 | ||
| Scheme 2 Photochemical dehalogenation of DDT. | ||
In conclusion, we have developed the first direct, visible light-induced photochemical reduction of native vitamin B12 to the supernucleophilic Co(I) form using an easily available and inexpensive MOF photocatalyst, MIL-125-NH2. Gratifyingly, the method does not require a prior reduction of vitamin B12 to the Co(II) form. Furthermore, the amount of vitamin B12 in these reactions is as low as 0.5 mol%, and the MOF co-catalyst can be recovered without substantial loss of activity. The utility of the system is demonstrated in the synthesis of pyrrolidine derivatives and the dehalogenation of DDT.
The authors thank the National Science Centre, Poland, for financial support (DG: OPUS UMO-2020/39/I/ST4/00405, MJC: OPUS UMO-2017/27/B/ST5/00941).
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Banerjee, Chemistry and Biochemistry of B12, Wiley, 1999 Search PubMed.
- T. Wdowik and D. Gryko, ACS Catal., 2022, 12, 6517–6531 CrossRef CAS.
- T. Koide, T. Ono, H. Shimakoshi and Y. Hisaeda, Coord. Chem. Rev., 2022, 470, 214690 CrossRef CAS.
- H. Shimakoshi, E. Sakumori, K. Kaneko and Y. Hisaeda, Chem. Lett., 2009, 38, 468–469 CrossRef CAS.
- M. Ociepa, A. J. Wierzba, J. Turkowska and D. Gryko, J. Am. Chem. Soc., 2020, 142, 5355–5361 CrossRef CAS PubMed.
- T. Troxler and R. Scheffold, Helv. Chim. Acta, 1994, 77, 1193–1202 CrossRef CAS.
- Y. Chen and X. P. Zhang, J. Org. Chem., 2004, 69, 2431–2435 CrossRef CAS.
- Z. Petrović, Z. Bugarčić, L. Marjanović and S. Konstantinović, J. Mol. Catal. A: Chem., 1999, 142, 393–395 CrossRef.
- H. Shimakoshi, M. Nishi, A. Tanaka, K. Chikama and Y. Hisaeda, Chem. Commun., 2011, 47, 6548–6550 RSC.
- S. Smoleń, A. Wincenciuk and D. Gryko, Synthesis, 2021, 1645–1653 CrossRef.
- M. Giedyk, K. Goliszewska and D. Gryko, Chem. Soc. Rev., 2015, 44, 3391–3404 RSC.
- D. Lexa and J.-M. Saveant, Acc. Chem. Res., 1983, 16, 235–243 CrossRef CAS.
- H. Tian, H. Shimakoshi, G. Park, S. Kim, Y. You and Y. Hisaeda, Dalton Trans., 2018, 47, 675–683 RSC.
- H. Shimakoshi and Y. Hisaeda, Angew. Chem., Int. Ed., 2015, 54, 15439–15443 CrossRef CAS PubMed.
- D. E. Yerien, A. Postigo, M. Baroncini and S. Barata-Vallejo, Green Chem., 2021, 23, 8147–8153 RSC.
- Q. Wang, Q. Gao, A. M. Al-Enizi, A. Nafady and S. Ma, Inorg. Chem. Front., 2020, 7, 300–339 RSC.
- J. Xu, H. Shimakoshi and Y. Hisaeda, J. Organomet. Chem., 2015, 782, 89–95 CrossRef CAS.
- S.-N. Zhao, G. Wang, D. Poelman and P. Van Der Voort, Molecules, 2018, 23, 2947–2969 CrossRef PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- J. Ding, M. Chen, X. Du, R. Shang, M. Xia, J. Hu and Q. Zhong, Catal. Lett., 2019, 149, 3287–3295 CrossRef CAS.
- X. Cheng, Y. Gu, X. Zhang, X. Dao, S. Wang, J. Ma, J. Zhao and W. Sun, Appl. Catal., B, 2021, 298, 120524 CrossRef CAS.
- F. Guo, M. Yang, R. Li, Z. He, Y. Wang and W. Sun, ACS Catal., 2022, 12, 9486–9493 CrossRef CAS.
- H. Huang, X. Wang, D. Philo, F. Ichihara, H. Song, Y. Li, D. Li, T. Qiu, S. Wang and J. Ye, Appl. Catal., B, 2020, 267, 118686 CrossRef CAS.
- H. Wang, X. Yuan, Y. Wu, G. Zeng, X. Chen, L. Leng, Z. Wu, L. Jiang and H. Li, J. Hazard. Mater., 2015, 286, 187–194 CrossRef CAS PubMed.
- R. R. Solís, A. Gomez-Aviles, C. Belver, J. J. Rodriguez and J. Bedia, J. Environ. Chem. Eng., 2021, 9, 106230 CrossRef.
- H.-T. N. Thi, K.-N. T. Thi, N. B. Hoang, B. T. Tran, T. S. Do, C. S. Phung and K.-O. N. Thi, Materials, 2021, 14, 7741–7753 CrossRef PubMed.
- J. Wang, A. S. Cherevan, C. Hannecart, S. Naghdi, S. P. Nandan, T. Gupta and D. Eder, Appl. Cat. B, 2021, 283, 119626 CrossRef CAS.
- D. Sun, L. Ye and Z. Li, Appl. Cat. B, 2015, 164, 428–432 CrossRef CAS.
- X. Tan, J. Zhang, J. Shi, X. Cheng, D. Tan, B. Zhang, L. Liu, F. Zhang, B. Han and L. Zheng, Sustainable Energy Fuels, 2020, 4, 2823–2830 RSC.
- K. Park and T. C. Brunold, J. Phys. Chem. B, 2013, 117, 5397–5410 CrossRef CAS PubMed.
- W. Peng, Y. Lin, Z. Wan, H. Ji, W. Ma and J. Zhao, Catal. Today, 2020, 340, 86–91 CrossRef CAS.
- M. A. Nasalevich, R. Becker, E. V. Ramos-Fernandez, S. Castellanos, S. L. Veber, M. V. Fedin, F. Kapteijn, J. N. H. Reek, J. I. van der Vlugt and J. Gascon, Energy Environ. Sci., 2015, 8, 364–375 RSC.
- T. Fujioka, T. Nakamura, H. Yorimitsu and K. Oshima, Org. Lett., 2002, 4, 2257–2259 CrossRef CAS PubMed.
- R. Scheffold, G. Rytz, L. Walder, R. Orlinski and Z. Chilmonczyk, Pure Appl. Chem., 1983, 55, 1791–1797 CrossRef CAS.
- M. A. Jabbar, H. Shimakoshi and Y. Hisaeda, Chem. Commun., 2007, 1653–1655 RSC.
- Y. Anai, K. Shichijo, M. Fujitsuka and H. Shimakoshi, Chem. Commun., 2020, 56, 11945–11948 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc02941g |
| This journal is © The Royal Society of Chemistry 2023 |