
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient light-driven hydrogen evolution utilizing porphyrin-based nanoparticles†
Vasilis
Nikolaou‡
 a,
Eleni
Agapaki‡
a,
Emmanouil
Nikoloudakis
a,
Katerina
Achilleos
a,
Kalliopi
Ladomenou
b,
Georgios
Charalambidis
a,
Evitina
Triantafyllou
a and
Athanassios G.
Coutsolelos
*ac
a,
Eleni
Agapaki‡
a,
Emmanouil
Nikoloudakis
a,
Katerina
Achilleos
a,
Kalliopi
Ladomenou
b,
Georgios
Charalambidis
a,
Evitina
Triantafyllou
a and
Athanassios G.
Coutsolelos
*ac
aLaboratory of Bioinorganic Chemistry, Department of Chemistry, University of Crete, Voutes Campus, 70013 Heraklion, Crete, Greece. E-mail: acoutsol@uoc.gr
bLaboratory of Inorganic Chemistry, Department of Chemistry, International Hellenic University, 65404 Kavala, Greece
cInstitute of Electronic Structure and Laser (IESL) Foundation for Research and Technology - Hellas (FORTH), Vassilika Vouton, 70013 Heraklion, Crete, Greece
First published on 22nd August 2023
Abstract
We developed dye-sensitized photocatalytic systems (DSPs) by utilizing porphyrins as a photosensitizer (PS) or as a photosensitizer–catalyst (PS/CAT) upon their chemisorption onto platinum-doped titanium dioxide nanoparticles (Pt-TiO2 NPs). The DSPs coated with Pt-Tc3CP (PS/CAT entity) exhibited a record-high stability (25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 TONs) and H2 evolution activity (707 mmol g−1 h−1) compared to similar DSPs in the literature.
500 TONs) and H2 evolution activity (707 mmol g−1 h−1) compared to similar DSPs in the literature.
The prolonged and continuous utilization of fossil fuels as primary energy sources generated significant environmental complications, since their combustion is responsible for the exponential increase of greenhouse gas emissions.1 Hence, it is of great importance to establish cost-competitive, low-carbon technologies aiming to develop sustainable systems based on renewable sources.2 Green hydrogen (H2) besides being a clean fuel is a chemical feedstock as well, which can be imperative for accomplishing multi-sector decarbonization.3 Light-driven H2 production is an auspicious method because an abundant and clean energy (solar) is used to achieve an energy-rich storable compound (H2) that can be used as a fuel.4
An efficient and facile approach toward light-driven H2 evolution is the development of dye-sensitized photocatalytic systems (DSPs). Indeed, DSPs have received considerable attention recently due to their durability and tunability.5,6 In such photocatalytic schemes, a photosensitizer (PS) and a catalyst (CAT) are immobilized onto titanium dioxide nanoparticles (TiO2 NPs) forming a heterogeneous photocatalyst. Using a light source, the PS absorbs photons and gets excited; subsequently, electrons are injected into the conduction band of TiO2. Finally, this flow of electrons reaches the CAT, which performs the reduction of H+ to H2 and the oxidized PS is regenerated using a sacrificial electron donor (SED).
A great number of different photosensitizers (PSs) and catalysts (CATs) have been utilized in TiO2-based DSPs over the last decade (Fig. 1a).6,7 In addition, by simply exchanging a molecular CAT with highly efficient Pt, researchers were able to develop schemes with greater stability and higher H2 evolution activity (Fig. 1b).8,9 There are various examples of porphyrinoids being utilized either as the PS or as the CAT in photocatalytic H2 evolution, mainly due to their straightforward structural modification, which enables them to adopt desirable chemical and physical properties.10,11 In spite of this fact though, only recently, our research group demonstrated a different approach (Fig. 1c), in which a single porphyrin (PS/CAT) was able to act as the light harvester and simultaneously as the catalyst.12 Thus, in this approach, there is only one component (PS/CAT) that drives the photocatalytic H2 evolution. Moving one step forward, herein we introduce an alternative approach, in which a PS/CAT derivative (a light harvester and a catalyst) is incorporated into Pt-TiO2 NPs. The Pt-TiO2 NPs act as a scaffold for the successful self-organization of the PS/CAT entities, an electron transport medium, and also as an additional photocatalyst. Thus, we combined metalated-porphyrin carboxylic acid derivatives with Pt-TiO2 NPs and successfully developed highly efficient DSPs for H2 evolution in aqueous media (Fig. 1d).
 | ||
| Fig. 1 The three established approaches in the literature to design DSPs for H2 evolution: (a) PS and CAT on TiO2 NPs, (b) PS onto PtTiO2 NPs, and (c) PS–CAT onto TiO2 NPs. (d) New approach: PS–CAT onto Pt-TiO2 NPs. | ||
We prepared two series of porphyrin derivatives metalated with zinc (Zn), palladium (Pd), and platinum (Pt) and introduced carboxylic acid units at their periphery as anchoring groups for their successful attachment onto Pt-TiO2 NPs. As illustrated in Fig. S1 (ESI†), the carboxylic acid units were introduced either on a three-carbon alkyl chain (c3) at the para-position of the phenyl ring (for the M-Tc3CP porphyrins) or at the para-position of the phenyl ring (for the derivatives M-TCP). The leading reasons to specifically modify these porphyrinoids in such a manner were derived from the results of our previous investigations.12 Namely, Pd-Tc3CP and Pt-Tc3CP were able to self-organize onto TiO2 NPs acting as both the PS and the CAT. In stark contrast, Zn-Tc3CP acted only as the PS, and as expected it did not produce any H2. Although these DSPs demonstrated great stability and efficiency, the best conditions for H2 evolution should be an organic/aqueous solvent mixture with 15% of triethanolamine as the SED. Thus, to explore their catalytic properties onto Pt-TiO2 DSPs, we prepared three metalated M-Tc3CP (with Zn, Pd, and Pt, Fig. S1, ESI†) together with their respective M-TCP derivatives. More importantly, all these porphyrin-based Pt-TiO2 DSPs operate in aqueous medium, rendering them as “greener” DSPs compared to the previous ones.12
The synthesis of M-Tc3CP was carried out following the procedures reported in the literature,12–14 whereas M-TCP derivatives were prepared according to the synthetic approach described in detail in the ESI† (Scheme S1 and Fig. S2–S13). The absorption spectra of M-TCP and M-Tc3CP in freshly distilled toluene/ethanol solutions (ratio 1:1) are illustrated in Fig. 2. Their absorption coefficient numbers and their absorption data (λmax of Soret and Q bands) are listed in Table S1 (ESI†). In all cases, typical absorption features for such metalated porphyrinoids15 are observed. The electrochemical properties of all M-Tc3Ps were investigated by means of cyclic voltammetry (Table S2, ESI†). The driving force regarding the electron injection from the porphyrins to TiO2 (ΔGinj), as well as their regeneration from the SED (ΔGreg) indicate that both processes are thermodynamically favorable (see the ESI† for details; Table S2).
 | ||
| Fig. 2 Extinction coefficient (ε) of (a) M-TCP and (b) M-c3TCP derivatives (M: Zn, Pd, or Pt) in freshly distilled toluene/ethanol (1:1 ratio) solutions. (c) Absorption spectra of Pt-Tc3CP (1.0 × 10−5 M) before and after chemisorption onto Pt-TiO2 NPs. | ||
The initial step for the light-driven H2 evolution experiment is the chemisorption of the complexes onto the Pt-TiO2 NPs. Various initial concentrations of metalated porphyrins were utilized in order to optimize the chemisorption of the porphyrins onto Pt-TiO2 NPs (see the ESI†). All the absorption spectra before and after their chemisorption are available in the ESI† (Fig. S14–S16) and the respective dye loadings (DLs) for each complex are listed in Tables S3–S8 (ESI†). In Fig. 2c, the absorption spectra of Pt-Tc3CP before and after chemisorption onto Pt-TiO2 NPs are presented. It is worth noting that by using this initial concentration (viz. 1.0 × 10−5 M), a quantitative loading of the porphyrin (DL = 100%) was achieved in all different cases.
In a typical H2 evolution experiment, 5 mg of the porphyrin-sensitized Pt-TiO2 NPs were dispersed in an aqueous solution (see the ESI† for details). The dispersions were prepared in a glass vial, sealed with a rubber septum, and irradiated using a 40 W light-emitting diode lamp (LED lamp, Fig. S17, ESI†). Upon photo-irradiation, all different metalated porphyrins (M-TCP and M-Tc3CP) demonstrated H2 production. The photocatalytic activity of the porphyrin-based DSPs was strongly dependent on the amount of the adsorbed porphyrin onto the TiO2 NPs (Tables S2–S7, ESI†). By considering the best photocatalytic data for each DSP (Table 1), we can safely conclude that all the M-Tc3TCP derivatives outperform their respective M-TCP ones, in terms of both stability (TONs) and efficiency (total H2 evolution, mmol g−1 h−1). Nevertheless, Zn-TCP and Zn-Tc3TCP demonstrated almost identical TONs (∼1100) and nearly the same amount of H2 evolution (∼400 mmol g−1 h−1). This indicates that the photocatalytic activity of Zn–porphyrin derivatives is not affected by the different anchoring groups (the rigid carboxylic acid group vs. flexible c3-carboxylic acid), confirming their sole role as PSs.
| Porphyrin | TONs vs. PSa | TONs vs. CATb | H2 evolution ratec |
|---|---|---|---|
| a TON in 24 hours vs. PS = porphyrin (see the ESI for details). b TON in 24 hours vs. CAT. In the cases of Zn-TCP and Zn-Tc3CP: CAT = PtTiO2. However, in the cases of Pd-TCP, Pd-Tc3CP, Pt-TCP, and Pt-Tc3CP: CAT = PtTiO2 and Pt- or Pd-porphyrin (see the ESI for details). c H2 evolution rate (mmol g−1 h−1) vs. total g of CAT in 24 hours (see the ESI for details). | |||
| Zn-TCP | 1192 | 1686 | 360 |
| Zn-Tc3CP | 1031 | 1972 | 421 |
| Pd-TCP | 1147 | 814 | 256 |
| Pd-Tc3CP | 7722 | 2097 | 593 |
| Pt-TCP | 2525 | 1772 | 378 |
| Pt-Tc3CP | 11607 |
1216 | 707 |
In contrast, both Pd-Tc3TCP and Pt-Tc3TCP exhibited superior TONs and H2 evolution rates compared to their counterparts Pd-TCP and Pt-TCP, respectively (Table 1). More specifically, Pd-Tc3CP achieved 7722 TONs and 593 mmol g−1 h−1 of H2 evolution, the values of which are significantly greater than those of Pd-TCP (1147 TONs and 256 mmol g−1 h−1 of H2, respectively). In a similar manner, nonetheless reaching even greater results, Pt-Tc3CP demonstrated 11607 TONs and 458 mmol g−1 h−1 of H2 evolution, outperforming Pt-TCP (2525 TONs and 378 mmol g−1 h−1 of H2). A possible explanation for the enhanced performance of the M-Tc3CP derivatives compared to that of the M-TCP ones is that the adsorption via four anchoring groups leads to a well-oriented self-organization of the M-Tc3CPs. This was indeed verified by scanning electron microscopy (SEM) experiments, which demonstrated that Pt-Tc3P undergoes a well-oriented self-organization in spherical nanostructures (Fig. S18b, ESI†). In contrast, both Zn-TCP (Fig. S18c, ESI†) and Pt-TCP (Fig. S18d, ESI†) self-aggregate on the top of the spherical Pt-TiO2 NPs (Fig. S18a, ESI†) In addition, the strong binding of Pt-Tc3CP is illustrated in Fig. S19 (ESI†), since the peaks correlated with the v(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and the v(C–O) stretching of the unbound carboxylic acid group at ∼1700 and 1200 cm−1, respectively disappeared in the FT-IR spectrum of the chemisorbed sample (Pt-Tc3CP@Pt-TiO2). In perfect agreement with our recent report, these catalytic results underline again the diverse behavior of both Pd- and Pt-porphyrins acting as PS/CAT entities, in contrast to the respective Zn-porphyrins, which act as PSs.12
O) and the v(C–O) stretching of the unbound carboxylic acid group at ∼1700 and 1200 cm−1, respectively disappeared in the FT-IR spectrum of the chemisorbed sample (Pt-Tc3CP@Pt-TiO2). In perfect agreement with our recent report, these catalytic results underline again the diverse behavior of both Pd- and Pt-porphyrins acting as PS/CAT entities, in contrast to the respective Zn-porphyrins, which act as PSs.12
In an effort to provide an even more fair assessment for the porphyrin-based DSPs developed herein, we explored their catalytic activity under the same dye loading (DL = 100%) by utilizing initial porphyrin solutions of 1.0 × 10−5 M in all cases (see Tables S3–S8, ESI†). As illustrated in Fig. 3, Pt-Tc3CP and Pd-Tc3CP reached 10819 and 7722 TONs and H2 evolution rates of 707 and 342 mmol g−1 h−1, respectively, outperforming their M-TCP counterparts and the respective Zn-porphyrinoinds. Similar to the behavior during the best performing photovoltaic experiments (Table 1), using these DSPs (with DL = 100%), the photocatalytic response of Zn-Tc3CP and Zn-TCP was yet again almost identical (∼1100 TONs and ∼50 mmol g−1 h−1 of H2). Furthermore, by comparing the M-TCP derivatives, evidently Pt-TCP (2018 TONs and 82 mmol g−1 h−1 of H2) is a better photocatalyst compared to Pd-TCP (1096 TONs and 49 mmol g−1 h−1 of H2). It may be well argued that Pd-TCP acts as a photosensitizer in this case, since the photocatalytic values are almost the same as the Zn–porphyrin-based DSPs (∼1100 TONs and ∼50 mmol g−1 h−1 of H2). In contrast, Pt-TCP reached almost double values concerning the TONs and the H2 evolution rates (2018 TONs and 82 mmol g−1 h−1, respectively) emphasizing once more the PS/CAT nature of the Pt–porphyrin-based DSPs.12
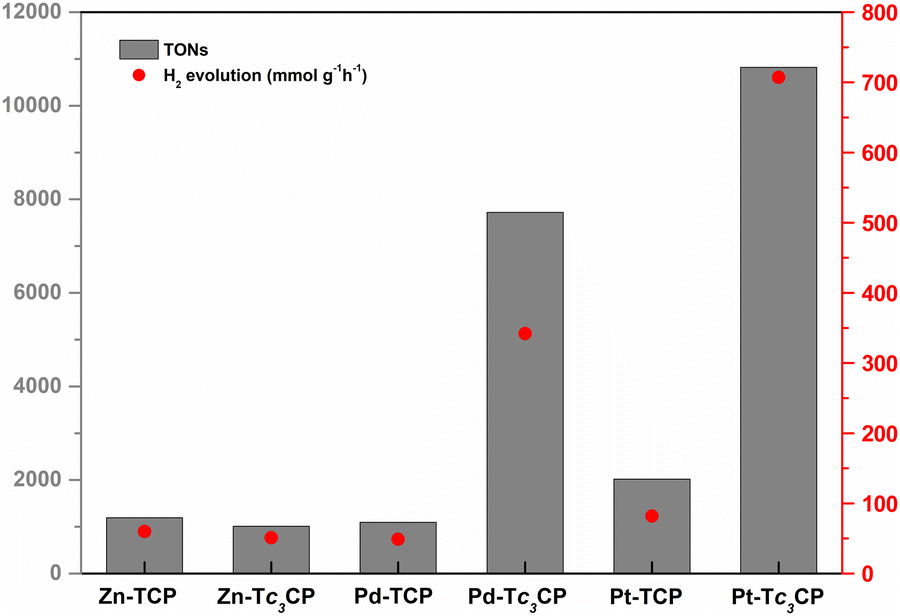 | ||
| Fig. 3 Comparison of all the M-TCP and M-c3TCP derivatives (M: Zn, Pd, or Pt) concerning their catalytic activity (TONs and H2 evolution rates). In all cases, the concentration of the initial porphyrin solution used for the chemisorption of the porphyrin was 1.0 × 10−5 M and the resulting dye-loading percentage was DL = 100%. | ||
To evaluate the stability of our best performing system (Pt-Tc3CP@PtTiO2), we performed long-term photocatalytic experiments. As illustrated in Fig. S20a (ESI†), the DSPs coated with Pt-Tc3CP exhibited great stability reaching 25500 TONs, and high H2 evolution activity (316 mmol g−1 h−1 of H2). Upon 48 h of continuous visible light irradiation, the H2 evolution was stopped and the DSPs reached a plateau. In an effort to investigate the reason for deactivation, in four different experiments, we added either (a) Pt-Tc3CP, (b) AA 1M, (c) Pt-TiO2, or (d) Pt-Tc3CP@Pt-TiO2; however, we did not observe any reactivation of the H2 production (Fig. S20b, ESI†). Only when both the photocatalytic NPs (Pt-Tc3CP@Pt-TiO2) and the SED (AA) were added, H2 production was detected. These experiments demonstrate that both the SED and the Pt-Tc3CP@Pt-TiO2 photocatalyst have been converted into non-active compounds/materials. Indeed, the absorption spectrum of the SED before and after the catalytic experiment significantly changes (Fig. S20c, ESI†). In contrast, the SEM experiments demonstrated that the morphology of Pt-TiO2 is not affected at all upon photocatalysis (Fig. S21, ESI†). More specifically, similar to our previous study,19Pt-Tc3CP exhibits a spherical self-organization on the Pt-TiO2 NPs which is retained even upon the 48 h of photocatalysis. Hence, we concluded that the deactivation of the Pt-Tc3CP@Pt-TiO2 system can be attributed to the degradation of the porphyrin and the SED, rendering them the limiting factors of our DSPs.
In addition, we evaluated the correlation between the amount of porphyrin and the H2 evolution rate for all M-Tc3CP and M-TCP (M: Zn, Pt, and Pd, Fig. S22, ESI†). Interestingly, for all M-TCP porphyrins and Zn-Tc3CP, a linear correlation between the amount of the porphyrin and the H2 evolution rate is observed. In particular, in all cases, the higher H2 evolution rate was detected for the greater porphyrin quantity (∼ 3.0 × 10−7 mol). In stark contrast, the respective experiments for Pt-Tc3CP and Pd-Tc3CP revealed that the highest H2 production rate is not associated with the greater amount of the porphyrin. In the case of Pd-Tc3CP, the highest H2 production rate of 1.5 × 10−7 mol was detected, whereas for the Pt-Tc3CP it was 3.0 × 10−8 mol of the porphyrin. These findings confirm once more the hypothesis that the enhanced catalytic efficiency of Pt-Tc3CP and Pd-Tc3CP can be accredited to the well-oriented self-organization of the porphyrinoids on the surface of the Pt-TiO2 performing as PS/CAT entities.
To sum up, we demonstrated that dispersion of porphyrin-based DSPs in an aqueous medium is an auspicious method to develop highly performing photocatalytic nanoparticles. Unlike the reported research works to date, the utilized porphyrins herein are able to act at the same time as a light harvester and a catalyst (PS/CAT). Hence, we introduced a more facile – in terms of preparation protocols – approach, compared to more complex structures (PS-dyads) or a combination of two entities (PS + CAT) that were reported in other studies.9–13,16–21 We prepared different metalated tetra-carboxylic acid porphyrins to explore the impact of two significant factors: (i) the different metals and (ii) the position of the anchoring group in light-driven H2 production. Interestingly, in contrast to Pd- and Pt-porphyrins, the catalytic properties of the respective Zn-porphyrins were not affected by the different anchoring groups. It is worth mentioning that the different positioning of the anchoring groups strongly impacted the H2 evolution of both Pd- and Pt-porphyrin-based DSPs. We can safely assume that the M-TCP derivatives use maximum two carboxylic acids to bound onto the NPs, whereas the chemisorption of M-Tc3CP can be achieved via four carboxylic acids.22–25 This multisided anchoring mode of M-Tc3CP derivatives resulted in their enhanced photocatalytic activity (Table 1).
In Table 2, a thorough comparison between our work and the highest performing porphyrin-based DSPs in the literature is given. In all these reports, a porphyrinoid or a porphyrin-based dyad was utilized as the PS and Pt-TiO2 NPs as the catalyst entity, and the photocatalytic reaction was performed in an aqueous solvent. Remarkably, the H2 evolution rate of our DSPs developed with Pt-Tc3CP is more than two times greater (707 mmol g−1 h−1) compared to other porphyrin-based DSPs reported in the literature (Table 1). In addition, besides the high efficiency, the developed DSPs herein exhibited a greater stability as well, reaching 25500 TONs. Overall, we showed stable and efficient DSPs for H2 evolution by utilizing PS–CAT derivatives in aqueous media. In the future, several aspects should be explored targeting the development of earth-abundant dispersions with high stability and efficiency.
| PS | SED | TONs | Irr. time | H2 evolutionb | Publication |
|---|---|---|---|---|---|
| a TON in 48 hours vs. PS = porphyrin (see the ESI for details). b H2 evolution rate (mmol g−1 h−1) vs. total g of CAT in 24 hours. CAT = PtTiO2 and Pt-Tc3CP (see the ESI for details). c The oxygenation of cyclohexene (C6H12) was studied instead of the presence of a SED. d In μmol g−1 h−1. | |||||
| Pt-Tc3CP | AA (1M) | 25500a |
24/48 | 707 | This work |
| YD2-o-C8 | AA (0.5M) | 11900 |
120 | 272 | 28 |
| BDP-Por-BDP(Im) | AA (1M) | 18600 |
72 | 225 | 31 |
| ZnP-dyad | AA (0.5M) | 12800 |
120 | 173 | 28 |
| LGtT | TEOA 20% | 14792 |
5 | 7.4 | 29 |
| LG-DtT | TEOA 20% | 13282 |
5 | 6.7 | 29 |
| LG-5 | TEOA 20% | 6582 | 5 | 3.3 | 29 |
| PdTHPP | TEOA 10% | 158 | 5 | 2.0 | 30 |
| ZnTHPP | TEOA 10% | 92 | 5 | 1.3 | 30 |
| THPP | TEOA 10% | 39 | 5 | 0.6 | 30 |
| SnTPyP | EDTA | 7.5 | 4 | 0.2 | 27 |
| Sn(IV)TCPP | C6H12c | — | 5 | 0.5d | 26 |
This research was financed by the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship, and Innovation, under the call RESEARCH-CREATE-INNOVATE (project code: T1EDK-01504). In addition, this research has been co-financed by the European Union and Greek national funds through the Regional Operational Program “Crete 2014-2020,” project code OPS:5029187. Moreover, the European Commission's Seventh Framework Program (FP7/2007-2013) under grant agreement no. 229927 (FP7- REGPOT-2008-1, Project BIO-SOLENUTI) and the Special Research Account of the University of Crete are gratefully acknowledged for the financial support of this research.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Du, D. Xiang, K. Zhou, L. Wang, J. Yu, H. Xia, L. Zhao, H. Liu and W. Zhou, Nano Energy, 2022, 104, 107875 CrossRef CAS.
- A. Rahman, O. Farrok and M. M. Haque, Renewable Sustainable Energy Rev., 2022, 161, 112279 CrossRef.
- S. Martinez-Villarreal, M. Kammoun and A. Richel, Curr. Opin. Green Sustainable Chem., 2023, 39, 100716 CrossRef CAS.
- J. Radhakrishnan, S. Ratna and K. Biswas, Inorg. Chem. Commun., 2022, 145, 109971 CrossRef CAS.
- G. Reginato, L. Zani, M. Calamante, A. Mordini and A. Dessì, Eur. J. Inorg. Chem., 2020, 899–917 CrossRef CAS.
- J. Willkomm, K. L. Orchard, A. Reynal, E. Pastor, J. R. Durrant and E. Reisner, Chem. Soc. Rev., 2016, 45, 9–23 RSC.
- S. Rajak, N.-N. Vu, P. Kaur, A. Duong and P. Nguyen-Tri, Coord. Chem. Rev., 2022, 456, 214375 CrossRef CAS.
- L. Zani, M. Melchionna, T. Montini and P. Fornasiero, J. Phys.: Energy, 2021, 3, 031001 CAS.
- J.-F. Huang, Y. Lei, T. Luo and J.-M. Liu, ChemSusChem, 2020, 13, 5863–5895 CrossRef CAS PubMed.
- E. Nikoloudakis, I. López-Duarte, G. Charalambidis, K. Ladomenou, M. Ince and A. G. Coutsolelos, Chem. Soc. Rev., 2022, 51, 6965–7045 RSC.
- J. S. O'Neill, L. Kearney, M. P. Brandon and M. T. Pryce, Coord. Chem. Rev., 2022, 467, 214599 CrossRef.
- V. Nikolaou, G. Charalambidis, K. Ladomenou, E. Nikoloudakis, C. Drivas, I. Vamvasakis, S. Panagiotakis, G. Landrou, E. Agapaki, C. Stangel, C. Henkel, J. Joseph, G. Armatas, M. Vasilopoulou, S. Kennou, D. M. Guldi and A. G. Coutsolelos, ChemSusChem, 2021, 14, 961–970 CrossRef CAS PubMed.
- I. D. Kostas, A. G. Coutsolelos, G. Charalambidis and A. Skondra, Tetrahedron Lett., 2007, 48, 6688–6691 CrossRef CAS.
- C. Stangel, D. Daphnomili, T. Lazarides, M. Drev, U. O. Krasovec and A. G. Coutsolelos, Polyhedron, 2013, 52, 1016–1023 CrossRef CAS.
- K. T. Weber, K. Karikis, M. D. Weber, P. B. Coto, A. Charisiadis, D. Charitaki, G. Charalambidis, P. Angaridis, A. G. Coutsolelos and R. D. Costa, Dalton Trans., 2016, 45, 13284–13288 RSC.
- K. Kurimoto, T. Yamazaki, Y. Suzuri, Y. Nabetani, S. Onuki, S. Takagi, T. Shimada, H. Tachibana and H. Inoue, Photochem. Photobiol. Sci., 2014, 13, 154–156 CrossRef CAS PubMed.
- E. Koposova, X. Liu, A. Pendin, B. Thiele, G. Shumilova, Y. Ermolenko, A. Offenhäusser and Y. Mourzina, J. Phys. Chem. C, 2016, 120, 13873–13890 CrossRef CAS.
- P.-Y. Ho, M. F. Mark, Y. Wang, S.-C. Yiu, W.-H. Yu, C.-L. Ho, D. W. McCamant, R. Eisenberg and S. Huang, ChemSusChem, 2018, 11, 2517–2528 CrossRef CAS.
- P. S. Gangadhar, S. Gonuguntla, S. Madanaboina, N. Islavath, U. Pal and L. Giribabu, J. Photochem. Photobiol., A, 2020, 392, 112408 CrossRef CAS.
- L. Y. Huang, J. F. Huang, Y. Lei, S. Qin and J. M. Liu, Catalysts, 2020, 10(6), 656 CrossRef CAS.
- V. Nikolaou, G. Charalambidis, G. Landrou, E. Nikoloudakis, A. Planchat, R. Tsalameni, K. Junghans, A. Kahnt, F. Odobel and A. G. Coutsolelos, Acs Appl. Energy Mater., 2021, 4, 10042–10049 CrossRef CAS.
- A. S. Hart, C. B. Kc, H. B. Gobeze, L. R. Sequeira and F. D’Souza, ACS Appl. Mater. Interfaces, 2013, 5, 5314–5323 CrossRef CAS PubMed.
- L. Zhang and J. M. Cole, ACS Appl. Mater. Interfaces, 2015, 7, 3427–3455 CrossRef CAS PubMed.
- K. Ladomenou, T. N. Kitsopoulos, G. D. Sharma and A. G. Coutsolelos, Rsc Adv., 2014, 4, 21379–21404 RSC.
- J. Rochford, D. Chu, A. Hagfeldt and E. Galoppini, J. Am. Chem. Soc., 2007, 129, 4655–4665 CrossRef CAS PubMed.
- K. Kurimoto, T. Yamazaki, Y. Suzuri, Y. Nabetani, S. Onuki, S. Takagi, T. Shimada, H. Tachibana and H. Inoue, Photochem. Photobiol. Sci., 2014, 13, 154–156 CrossRef CAS PubMed.
- E. Koposova, X. Liu, A. Pendin, B. Thiele, G. Shumilova, Y. Ermolenko, A. Offenhäusser and Y. Mourzina, J. Phys. Chem. C, 2016, 120, 13873–13890 CrossRef CAS.
- P.-Y. Ho, M. F. Mark, Y. Wang, S.-C. Yiu, W.-H. Yu, C.-L. Ho, D. W. McCamant, R. Eisenberg and S. Huang, ChemSusChem, 2018, 11, 2517–2528 CrossRef CAS PubMed.
- P. S. Gangadhar, S. Gonuguntla, S. Madanaboina, N. Islavath, U. Pal and L. Giribabu, J. Photochem. Photobiol., A, 2020, 392, 112408 CrossRef CAS.
- L. Y. Huang, J. F. Huang, Y. Lei, S. Qin and J. M. Liu, Catalysts, 2020, 10, 656 CrossRef CAS.
- V. Nikolaou, G. Charalambidis, G. Landrou, E. Nikoloudakis, A. Planchat, R. Tsalameni, K. Junghans, A. Kahnt, F. Odobel and A. G. Coutsolelos, ACS Appl. Energy Mater., 2021, 4, 10042–10049 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, synthesis and characterization, adsorption studies and photocatalysis details. See DOI: https://doi.org/10.1039/d3cc02922k |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |