
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceXanthine derivatives inhibit FTO in an L-ascorbic acid-dependent manner†
Kamui
Tanaka
 a,
Akiyo
Suda
a,
Motonari
Uesugi
abc,
Shiroh
Futaki
a and
Miki
Imanishi
*a
a,
Akiyo
Suda
a,
Motonari
Uesugi
abc,
Shiroh
Futaki
a and
Miki
Imanishi
*a
aInstitute for Chemical Research, Uji, Kyoto 611-0011, Japan. E-mail: imiki@scl.kyoto-u.ac.jp
bInstitute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University, Uji, Kyoto 611-0011, Japan
cSchool of Pharmacy, Fudan University, Shanghai 201203, People's Republic of China
First published on 21st August 2023
Abstract
Xanthine derivatives were identified as inhibitors of the N6-methyladenosine (m6A) demethylase activity of fat-mass-and-obesity-associated protein (FTO) by activity-based high-throughput screening using the m6A-sensitive ribonuclease MazF. Pentoxifylline exhibited L-ascorbic acid concentration-dependent inhibitory activity against FTO, an unprecedented mode of inhibition, indicating that L-ascorbic acid is a promising key for designing FTO-specific inhibitors.
N 6-Methyladenosine (m6A) is the most abundant RNA modification in mRNAs and ncRNAs. The methyl modification is installed by the writer complex and removed by eraser enzymes. To date, two RNA demethylases, fat-mass-and-obesity-associated protein (FTO) and alkB homolog 5 (ALKBH5), have been identified as m6A erasers. Through its association with m6A reader proteins, m6A modification plays a vital role in gene expression by regulating mRNA stability, localization, translation, and splicing.1–3 Thus, dysregulation of m6A erasers leads to abnormal gene expression resulting in various m6A-driven diseases. FTO has been reported to promote tumorigenesis such as VHL-deficient clear cell renal cell carcinoma (ccRCC) tumours4 and acute myeloid leukemia (AML).5 Therefore, small-molecule FTO modulators are promising for anticancer therapy.6,7
However, the development of FTO-selective inhibitors poses a substantial challenge due to the fact that both FTO and ALKBH5 are members of the Fe2+- and α-ketoglutarate (αKG)-dependent AlkB dioxygenase family, sharing a jelly roll motif as the catalytic domain for demethylating m6A.8,9 In fact, IOX-3, an αKG-competitive FTO inhibitor, is also known to inhibit αKG-dependent HIF-prolyl hydroxylases.10 The first FTO-directed inhibitor, rhein, also inhibits the m6A demethylation activity of ALKBH5.11 Recent efforts in the structure-based virtual screening for FTO active sites have led to the identification of FTO-specific small molecules.12–15 However, knowledge based on inhibitory modes and chemical backbones is still limited. A wide variety of compounds with novel binding sites or backbones offers opportunities to understand the protein function, which would aid in the elucidation of the FTO-dysregulated diseases. Therefore, the development of activity-based screening assays is required to identify FTO inhibitors with novel inhibitory mechanisms that are not restricted to known binding pockets.
High-performance liquid chromatography (HPLC) and mass spectrometry (MS) (HPLC-MS/MS)-based quantification of non-methylated adenosine versus m6A has been widely used as a conventional method for evaluating the demethylation activity of eraser enzymes.16 This method is highly quantitative but not adequate for high-throughput screening due to the time-consuming procedures and large sample requirements. A single-quantum-dot (QD)-based fluorescence resonance energy transfer (FRET) sensor using a methyl-sensitive restriction enzyme, DpnII, allows for the highly sensitive detection of FTO demethylation with low sample consumption.17 However, it uses DNA rather than RNA as the substrate and involves a multi-step procedure. Fluorescent RNA probes dependent on the methylation state of adenosines facilitate the activity-based high-throughput screening,18,19 even though the m6A-containing RNA sequences that form the probe structure are not consensus methylation sequences.
Here, the MazF endoribonuclease was utilized to perform activity-based high-throughput screening of FTO-selective inhibitors. We previously found that MazF, which cleaves single-stranded RNA at ACA sequences, does not cleave (m6A)CA sequences.20 Its applicability in monitoring FTO activity has been previously demonstrated.20–22 A 13-mer ssDNA/RNA chimera probe (5′ FAM-CAT r(GGm6ACA) TATGT-3′ BHQ-1) containing a typical methylation sequence, GG(m6A)CA, and labeled with a fluorescent dye and quencher at both ends, was used for the demethylation substrate by m6A demethylases. The demethylation activity of FTO and ALKBH5 can be evaluated based on increased fluorescence intensity, which is observed after MazF treatment, as it cleaves the demethylated fraction of the FRET probe, leading to the separation of the fluorophore and the quencher (Fig. 1).20 To confirm its validity of this system as a high-throughput screening system, a Z′-factor in the presence of 400 nM FTO was calculated to be 0.67, which exceeded the valid index of 0.5 (Fig. S1, ESI†).23
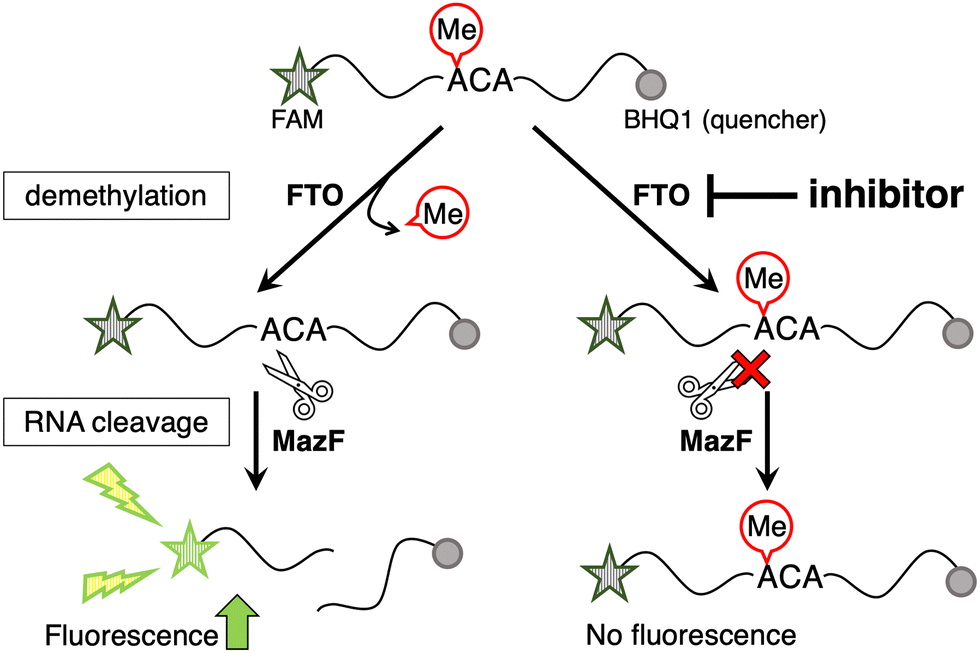 | ||
| Fig. 1 Principle of the identification of FTO inhibitors using activity-based high-throughput screening with m6A-sensitive ribonuclease, MazF. The self-quenched dual-labeled FRET probe (ssDNA/RNA chimera probe; 5′ FAM-CAT r(GGm6ACA) TATGT-3′ BHQ-1) was used as the substrate for FTO. Only demethylated probes are cleaved by MazF, resulting in increased fluorescence. Inhibition of the demethylation activity of FTO is reflected by a decrease in the fluorescence intensity of MazF-treated samples. | ||
Chemical libraries containing bioactive lipids, endocannabinoids, and neurotransmitters were screened, because FTO regulates adipogenesis24 and neurogenesis.25 Among approximately 400 bio-active compounds, four compounds (1; lisofylline (LSF), 2; 8-(p-sulfophenyl)theophylline, 3; 3-isobutyl-1-methylxanthine, and 4; 3,7-dimethyl-1-propargylxanthine) inhibited the demethylation activity of FTO at IC50 values <50 μM without inhibiting either the demethylation activity of ALKBH5 (Fig. 2 and Fig. S2, ESI†) or the RNA cleavage activity of MazF. Interestingly, these hit compounds are caffeine derivatives with a xanthine backbone, which is structurally similar to the adenine base but different from reported FTO inhibitors. The simplest xanthine derivatives, 9; theophylline and 10; caffeine, did not inhibit demethylation by FTO (Fig. 2). Among the hit compounds, compound 1, which has a 5-hydroxyhexyl group at the N1 position, showed the lowest IC50 value against FTO (IC50 = 2.2 μM) demonstrating the importance of the alkyl chain at the N1 position. Compound 1 and its analogs (5 and 6), with 5-oxohexyl and hexyl groups at the N1 position, respectively, also inhibited FTO with IC50 values of 4.1 and 1.4 μM, respectively. Further, 7, with a heptyl chain at position N1 instead of the hexyl chain of 6 showed a similar IC50 for FTO (IC50 = 1.5 μM). In contrast, 8, which has a propyl chain at N7 position instead of a methyl chain as in 5, showed no significant FTO inhibitory activity (Fig. 2, and Fig. S2, ESI†). These results indicated the importance of a hexyl or heptyl moiety at the N1 position and a methyl moiety at the N7 position of the xanthine backbone for the inhibitory effects on FTO. Considering the similar IC50 values of 1, 5, 6 and 7 regardless of the functional groups at the N1 position, it is plausible that the hydrophobic alkyl chain at the N1 position plays a key role in FTO inhibition. Compound 1 and its analogs (5 and 6) were also confirmed to inhibit the demethylation activity of FTO using gel electrophoresis of MazF-treated RNA oligonucleotides (Fig. S3A, ESI†). In addition, DpnII-dependent N6-methyl-deoxyadenosine (m6dA) demethylation assay11,26 showed that these compounds also inhibited FTO's ability to demethylate m6dA (Fig. S3B, ESI†). The results indicated that their inhibitory effects on FTO were not dependent on the evaluation method used for screening.
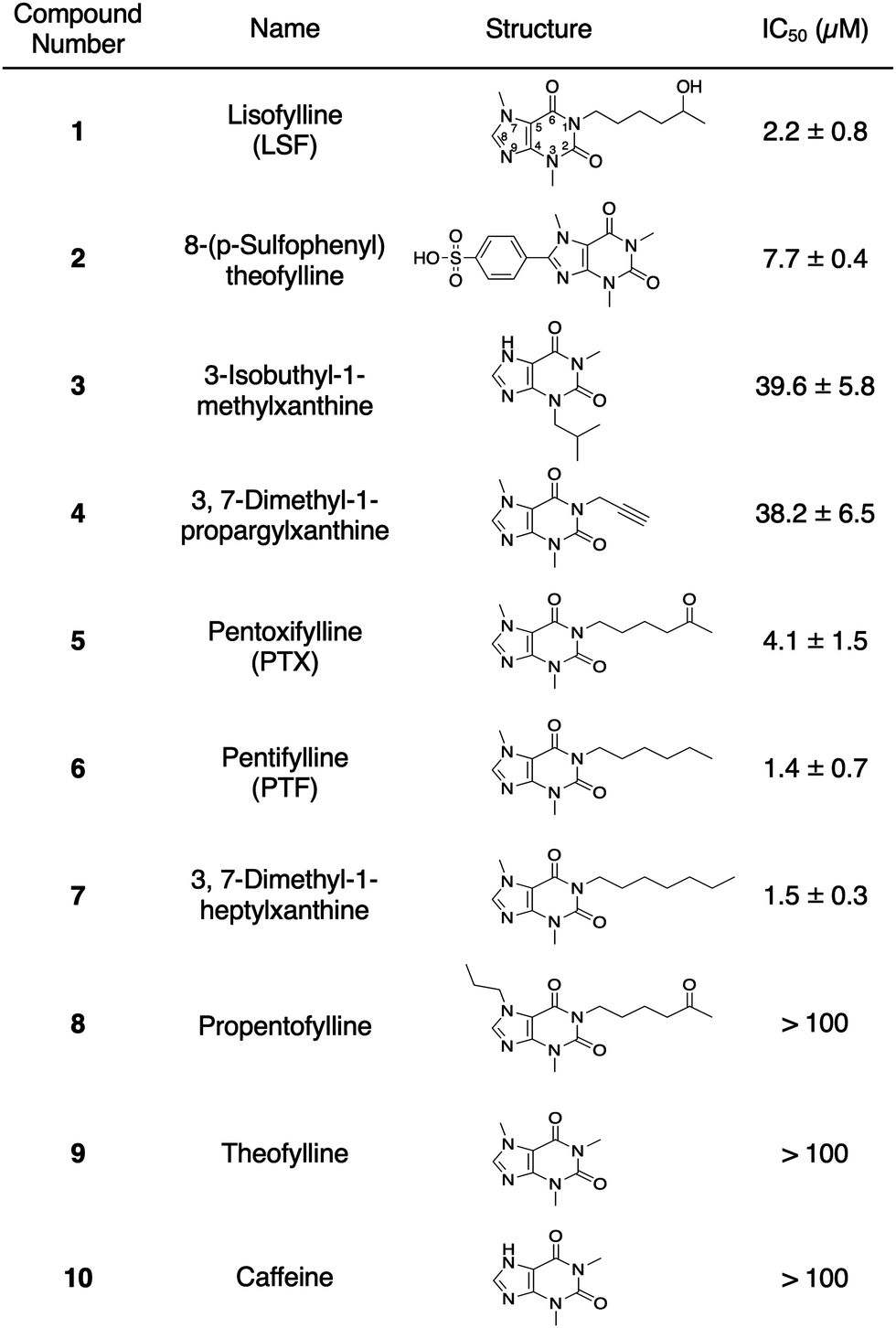 | ||
| Fig. 2 IC50 values of hit compounds (1–4) and their related compounds. | ||
The specificity of 1 and its analogs (5 and 6) for FTO was demonstrated by examining their inhibitory effects on other related enzymes. ALKBH5 and ten-eleven translocation methylcytosine dioxygenase 1 (TET1) are also Fe2+- and αKG-dependent dioxygenases and demethylate m6A and 5-methyl cytosine (5mC), respectively. These compounds at 100 μM, did not inhibit the demethylation activities of both ALKBH5 and TET1, whereas the m6A demethylation activity of FTO was completely inhibited (Fig. 3). Furthermore, they did not inhibit the methylation activity of m6A methyltransferase METTL3/14 (Fig. S2, ESI†), confirming the specificity of compounds 1, 5, and 6 for FTO over ALKBH5, TET1, and METTL3/14. Utilizing its solubility in water, 5 was selected as a representative analog of 1 for subsequent examination of the relevant inhibitory mechanisms.
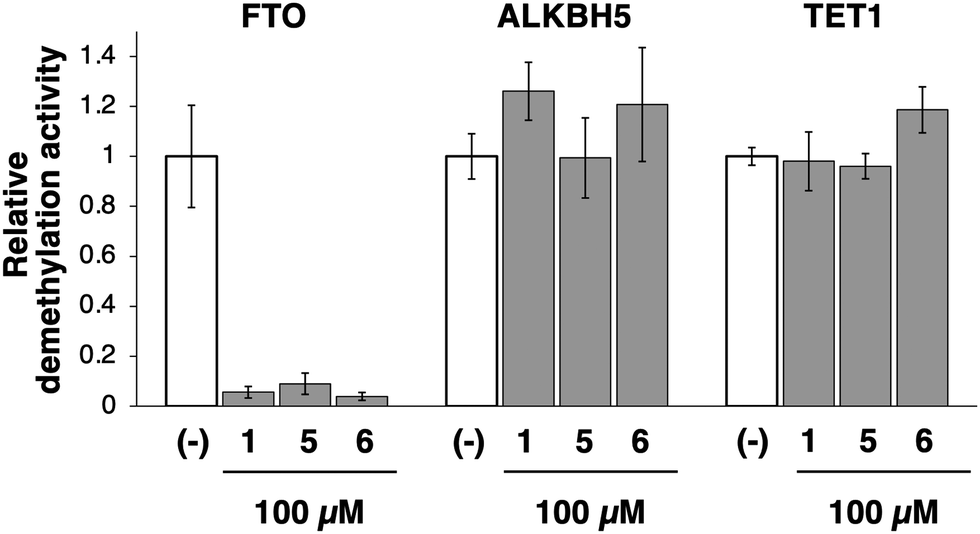 | ||
| Fig. 3 Specificity of compounds 1, 5, and 6 for FTO over ALKBH5 and TET1. The m6A demethylation activity of FTO and ALKBH5 was measured using a FRET-based MazF cleavage assay. The demethylation activity of TET1 was measured using ELISA with methylated plasmid DNA. | ||
Due to the xanthine backbone, which is common with purine bases, the effect of 5 on the interaction between FTO and the substrate RNA was examined using a fluorescence polarization (FP) assay (Fig. 4). No significant changes in the FP of the labeled RNA with FTO were observed in the presence of increasing amounts of 5. In contrast, a known substrate competitor of FTO, meclofenamic acid (MA),26 clearly decreased the molecular polarization of fluorescently labeled RNA in a concentration-dependent manner. These results demonstrated that 5 did not inhibit the interaction between the substrate RNA oligonucleotides and FTO. Further, the IC50 value of 5 did not correlate with αKG or Fe2+ concentrations (Fig. S4, ESI†). These results showed that compound 5 did not compete with the substrate RNA, αKG, or Fe2+.
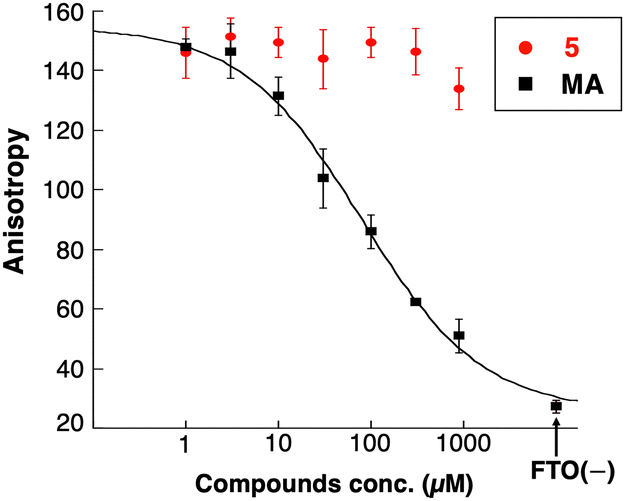 | ||
| Fig. 4 Compound 5 does not interfere with the interaction between FTO and substrate RNA. Fluorescence polarization of 20 nM of FAM-labeled RNA oligo mixed with 1 μM of FTO in the presence of 1–1000 μM of 5 or meclofenamic acid (MA) was measured. MA, a known FTO inhibitor, was used as a control to inhibit the RNA binding of FTO. | ||
Surprisingly, the IC50 value of compound 5 was significantly affected by the concentration of L-ascorbic acid. Specifically, the inhibitory effect of 5 became weaker as the concentration of L-ascorbic acid increased (Fig. 5). To confirm whether 5 competes with L-ascorbic acid, the effects of 5 were examined on the direct interaction between L-ascorbic acid and FTO using tryptophan fluorescence quenching.27 As a result, the binding affinity of L-ascorbic acid to FTO remained unchanged in the absence and presence of 5 at 50 μM, which mostly suppressed the demethylation activity of FTO (Fig. 6 and Fig. S5, ESI†). This result eliminates the possibility of the competition between 5 and L-ascorbic acid for binding with FTO.
 | ||
| Fig. 5 Inhibitory effect of 5 was affected by the concentration of L-ascorbic acid (L-AA). (A) Inhibitory curves of 5 against FTO in the presence of 0.3 mM (blue circle), 1 mM (black square), and 3 mM of L-ascorbic acid (red diamond). (B) IC50 values of 5, depending on the L-ascorbic acid concentration. | ||
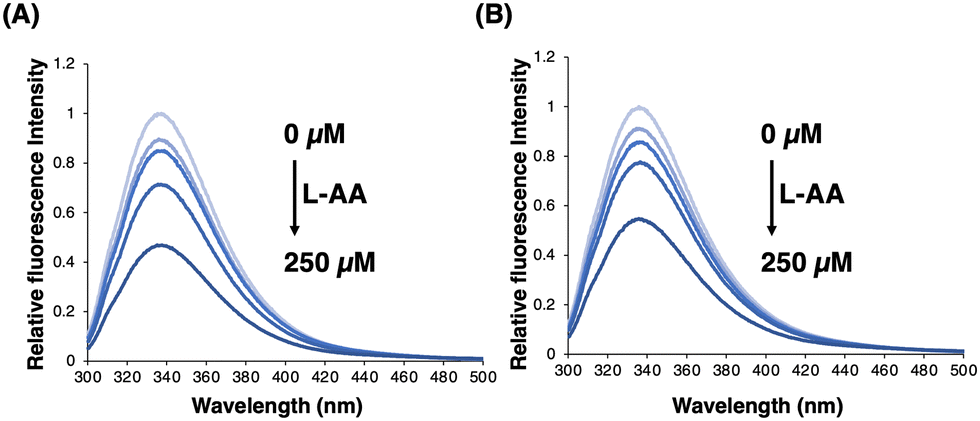 | ||
| Fig. 6 Direct binding of L-ascorbic acid (L-AA) to FTO. The fluorescence intensity of tryptophan was measured (ex: 280 nm). FTO was mixed with 0–250 μM L-AA in the absence (A) or presence (B) of 50 μM of compound 5. | ||
Moreover, although L-ascorbic acid has been regarded as a reducing agent, none of the other representative reducing agents, such as nicotinamide adenine dinucleotide phosphate (NADPH), dithiothreitol (DTT), tris(2-carboxyethyl)phosphine (TCEP), glutathione, gallic acid, tocopherol, or sodium borohydride (NaBH4), could replace L-ascorbic acid (Fig. S6, ESI†). This result highlights the importance of the direct interaction between L-ascorbic acid and FTO in the m6A demethylation reaction, rather than the reducing ability of L-ascorbic acid.
The exact mode of action of xanthine derivatives remains unclear. Nuclear magnetic resonance (NMR)-based structural analysis of apo FTO revealed dynamic fluctuations in the structure and the conformational variability of the domain–domain interface of FTO, which were not apparent from the crystallographic structures of holo FTO.28 We therefore hypothesize that compound 5 would interfere with these structural changes. The NMR study also indicated the presence of unknown druggable surface pockets apart from the catalytic core.28 Since 5 exhibited L-ascorbic acid-dependent inhibition of FTO without competing with L-ascorbic acid for binding sites, it is implied that 5 is an allosteric FTO inhibitor.
In this study, through the unique activity-based high-throughput screening method using MazF, xanthine derivatives, whose backbones differed from those of known FTO inhibitors, were identified as specific inhibitors of FTO. Surprisingly, the inhibition was modulated by L-ascorbic acid, a phenomenon that has not been previously reported in the literature. However, more research is needed to determine how L-ascorbic acid affects the demethylation process of FTO. Thus far, no reports have referred to inhibitors influenced by L-ascorbic acid. MO-I-500, an ascorbic acid mimic, originally designed to screen prolyl-4-hydroxylase 2 (PHD2) inhibitors, has been shown to inhibit FTO activity.29 However, the binding pocket of the hit compound is estimated to be at the αKG-binding site of FTO, and its relevance to ascorbic acid remains unclear. Further structural and functional analysis of FTO with L-ascorbic acid will reveal the contribution of L-ascorbic acid to the FTO activity and ultimately, the multifaceted function of FTO.
In comparison to static structure-based virtual screening, the activity-based approach offers a promising avenue for the discovery of novel FTO inhibitors with new backbones and mechanisms. These results have significant implications for understanding the regulatory mechanisms of FTO and the design of FTO-specific inhibitors, which have significant potential for anticancer treatment.
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI [19H02850, 21H05110, 22H02210 to M. I.].
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. C. He and C. He, EMBO J., 2021, 40, e105977 CrossRef CAS PubMed.
- H. Shi, J. Wei and C. He, Mol. Cell, 2019, 74, 640–650 CrossRef CAS PubMed.
- S. Zaccara, R. J. Ries and S. R. Jaffrey, Nat. Rev. Mol. Cell Biol., 2019, 20, 608–624 CrossRef CAS PubMed.
- Y. Xiao, K. N. Thakkar, H. Zhao, J. Broughton, Y. Li, J. A. Seoane, A. N. Diep, T. J. Metzner, R. von Eyben, D. L. Dill, J. D. Brooks, C. Curtis, J. T. Leppert, J. Ye, D. M. Peehl, A. J. Giaccia, S. Sinha and E. B. Rankin, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 21441–21449 CrossRef CAS PubMed.
- Z. Li, H. Weng, R. Su, X. Weng, Z. Zuo, C. Li, H. Huang, S. Nachtergaele, L. Dong, C. Hu, X. Qin, L. Tang, Y. Wang, G. M. Hong, H. Huang, X. Wang, P. Chen, S. Gurbuxani, S. Arnovitz, Y. Li, S. Li, J. Strong, M. B. Neilly, R. A. Larson, X. Jiang, P. Zhang, J. Jin, C. He and J. Chen, Cancer Cell, 2017, 31, 127–141 CrossRef PubMed.
- L. L. Zhou, H. Xu, Y. Huang and C. G. Yang, RSC Chem. Biol., 2021, 2, 1352–1369 RSC.
- S. Huff, I. R. Kummetha, L. Zhang, L. Wang, W. Bray, J. Yin, V. Kelley, Y. Wang and T. M. Rana, J. Med. Chem., 2022, 65, 10920–10937 CrossRef CAS PubMed.
- G. Jia, Y. Fu, X. Zhao, Q. Dai, G. Zheng, Y. Yang, C. Yi, T. Lindahl, T. Pan, Y. G. Yang and C. He, Nat. Chem. Biol., 2011, 7, 885–887 CrossRef CAS PubMed.
- G. Zheng, J. A. Dahl, Y. Niu, P. Fedorcsak, C. M. Huang, C. J. Li, C. B. Vågbø, Y. Shi, W. L. Wang, S. H. Song, Z. Lu, R. P. G. Bosmans, Q. Dai, Y. J. Hao, X. Yang, W. M. Zhao, W. M. Tong, X. J. Wang, F. Bogdan, K. Furu, Y. Fu, G. Jia, X. Zhao, J. Liu, H. E. Krokan, A. Klungland, Y. G. Yang and C. He, Mol. Cell, 2013, 49, 18–29 CrossRef CAS PubMed.
- W. Aik, M. Demetriades, M. K. Hamdan, E. A. Bagg, K. K. Yeoh, C. Lejeune, Z. Zhang, M. A. McDonough and C. J. Schofield, J. Med. Chem., 2013, 56, 3680–3688 CrossRef CAS PubMed.
- B. Chen, F. Ye, L. Yu, G. Jia, X. Huang, X. Zhang, S. Peng, K. Chen, M. Wang, S. Gong, R. Zhang, J. Yin, H. Li, Y. Yang, H. Liu, J. Zhang, H. Zhang, A. Zhang, H. Jiang, C. Luo and C. G. Yang, J. Am. Chem. Soc., 2012, 134, 17963–17971 CrossRef CAS PubMed.
- S. Peng, W. Xiao, D. Ju, B. Sun, N. Hou, Q. Liu, Y. Wang, H. Zhao, C. Gao, S. Zhang, R. Cao, P. Li, H. Huang, Y. Ma, Y. Wang, W. Lai, Z. Ma, W. Zhang, S. Huang, H. Wang, Z. Zhang, L. Zhao, T. Cai, Y. L. Zhao, F. Wang, Y. Nie, G. Zhi, Y. G. Yang, E. E. Zhang and N. Huang, Sci. Transl. Med., 2019, 11, eaau7116 CrossRef PubMed.
- M. Prakash, Y. Itoh, Y. Fujiwara, Y. Takahashi, Y. Takada, P. Mellini, E. E. Elboray, M. Terao, Y. Yamashita, C. Yamamoto, T. Yamaguchi, M. Kotoku, Y. Kitao, R. Singh, R. Roy, S. Obika, M. Oba, D. O. Wang and T. Suzuki, J. Med. Chem., 2021, 64, 15810–15824 CrossRef CAS PubMed.
- R. Su, L. Dong, Y. Li, M. Gao, L. Han, M. Wunderlich, X. Deng, H. Li, Y. Huang, L. Gao, C. Li, Z. Zhao, S. Robinson, B. Tan, Y. Qing, X. Qin, E. Prince, J. Xie, H. Qin, W. Li, C. Shen, J. Sun, P. Kulkarni, H. Weng, H. Huang, Z. Chen, B. Zhang, X. Wu, M. J. Olsen, M. Müschen, G. Marcucci, R. Salgia, L. Li, A. T. Fathi, Z. Li, J. C. Mulloy, M. Wei, D. Horne and J. Chen, Cancer Cell, 2020, 38, 79–96 CrossRef CAS PubMed e11.
- R. Wang, Z. Han, B. Liu, B. Zhou, N. Wang, Q. Jiang, Y. Qiao, C. Song, J. Chai and J. Chang, Mol. Pharm., 2018, 15, 4092–4098 CrossRef CAS PubMed.
- G. Jia, C. G. Yang, S. Yang, X. Jian, C. Yi, Z. Zhou and C. He, FEBS Lett., 2008, 582, 3313–3319 CrossRef CAS PubMed.
- Y. Zhang, Q. N. Li, K. Zhou, Q. Xu and C. Y. Zhang, Anal. Chem., 2020, 92, 13936–13944 CrossRef CAS PubMed.
- A. Cheong, J. J. A. Low, A. Lim, P. M. Yen and E. C. Y. Woon, Chem. Sci., 2018, 9, 7174–7185 RSC.
- N. Svensen and S. R. Jaffrey, Cell Chem. Biol., 2016, 23, 415–425 CrossRef PubMed.
- M. Imanishi, S. Tsuji, A. Suda and S. Futaki, Chem. Commun., 2017, 53, 12930–12933 RSC.
- K. Shinoda, A. Suda, K. Otonari, S. Futaki and M. Imanishi, Chem. Commun., 2020, 56, 1365–1368 RSC.
- X. Han, Y. Li, Z. Y. Wang, L. Z. Liu, J. G. Qiu, B. J. Liu and C. Y. Zhang, Chem. Commun., 2022, 58, 1565–1568 RSC.
- J. H. Zhang, T. D. Chung and K. R. Oldenburg, J. Biomol. Screen., 1999, 4, 67–73 CrossRef PubMed.
- X. Zhao, Y. Yang, B. F. Sun, Y. Shi, X. Yang, W. Xiao, Y. J. Hao, X. L. Ping, Y. S. Chen, W. J. Wang, K. X. Jin, X. Wang, C. M. Huang, Y. Fu, X. M. Ge, S. H. Song, H. S. Jeong, H. Yanagisawa, Y. Niu, G. F. Jia, W. Wu, W. M. Tong, A. Okamoto, C. He, J. M. Rendtlew Danielsen, X. J. Wang and Y. G. Yang, Cell Res., 2014, 24, 1403–1419 CrossRef CAS PubMed.
- L. Li, L. Zang, F. Zhang, J. Chen, H. Shen, L. Shu, F. Liang, C. Feng, D. Chen, H. Tao, T. Xu, Z. Li, Y. Kang, H. Wu, L. Tang, P. Zhang, P. Jin, Q. Shu and X. Li, Hum. Mol. Genet., 2017, 26, 2398–2411 CrossRef CAS PubMed.
- Y. Huang, J. Yan, Q. Li, J. Li, S. Gong, H. Zhou, J. Gan, H. Jiang, G. F. Jia, C. Luo and C. G. Yang, Nucleic Acids Res., 2015, 43, 373–384 CrossRef CAS PubMed.
- R. Yin, S. Q. Mao, B. Zhao, Z. Chong, Y. Yang, C. Zhao, D. Zhang, H. Huang, J. Gao, Z. Li, Y. Jiao, C. Li, S. Liu, D. Wu, W. Gu, Y. G. Yang, G. L. Xu and H. Wang, J. Am. Chem. Soc., 2013, 135, 10396–10403 CrossRef CAS PubMed.
- B. Khatiwada, T. T. Nguyen, J. A. Purslow and V. Venditti, J. Biol. Chem., 2022, 298, 101907 CrossRef CAS PubMed.
- G. Zheng, T. Cox, L. Tribbey, G. Z. Wang, P. Iacoban, M. E. Booher, G. J. Gabriel, L. Zhou, N. Bae, J. Rowles, C. He and M. J. Olsen, ACS Chem. Neurosci., 2014, 5, 658–665 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc02484a |
| This journal is © The Royal Society of Chemistry 2023 |