
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnbiased in silico design of pH-sensitive tetrapeptides†
Yue
Hu‡
 a,
Federica
Rigoldi‡
a,
Hui
Sun
a,
Alfonso
Gautieri
*b and
Benedetto
Marelli
*a
a,
Federica
Rigoldi‡
a,
Hui
Sun
a,
Alfonso
Gautieri
*b and
Benedetto
Marelli
*a
aCivil and Environmental Engineering, Massachusetts Institute of Technology, Cambridge, USA. E-mail: alfonso.gautieri@polimi.it; bmarelli@mit.edu
bBiomolecular Engineering Lab, Dipartimento di Elettronica, Informazione e Bioingegneria, Politecnico di Milano, Milano, Italy
First published on 31st July 2023
Abstract
We used coarse-grain molecular dynamics simulations to screen all possible histidine-bearing tetrapeptide sequences, finding novel peptide sequences with pH-tunable assembly properties. These tetrapeptides could be used for various biological applications, such as triggered delivery of bioactive molecules.
Oligopeptides are short peptides consisting of two to twenty amino acids (AA) that can spontaneously fold and assemble through a combination of hydrogen bonds and π–π interactions to form functional nanostructures.1 Oligopeptides can be used both as models to study protein behavior and as biomaterials with pre-defined structure–function relationship by selecting amino acid composition. Clinically, the study of oligopeptide aggregation is associated with formation of β-sheet-dominated structures called amyloids that are linked to diseases such as Alzheimer's and Parkinson's.2–4 From an engineering perspective, oligopeptides folding and assembly can be designed to fabricate fibers, tubes, nanosheets, pallets, gels, vesicles, and nanoparticles,5 with applications in biomedicine, food science, regenerative medicine, and biosensing.6–10 Peptide-based biomaterials offer in fact the opportunity to combine the simplicity of small biomolecules with the functionality of proteins. The design of oligopeptides that assemble in nanostructured materials often follows principles of bioinspiration and rational design. Most of the short (i.e. <5 AA) oligopeptides-based biomaterials found in literature are directly derived from AA motif with biological relevance (e.g. DFNKF, KLVFF) and/or are composed of hydrophobic AA (e.g. FFF, VYV) to drive self-assembly in water.11 The combination of bioinspiration with rational design, despite successful, has limited the design of oligopeptides to few sequences tested experimentally, when compared to the xn (where x = 20 AA and n = number of AA in the oligopeptide) theoretical possibilities, and biased the AA choice to impart low solubility. Such restrictions have strongly limited the discovery of new AA sequences.
As an alternative route, in silico tools can be used to predict oligopeptides’ properties, accelerating their design into biomaterials. Machine learning (ML) algorithms such as TorchMD, convolutional neural networks, and deep neural networks are also used to quickly model and predict peptides’ folding, energies, and reaction pathways, but the limited accuracy and completeness of high-quality training data still limit the resolution of predictive results of ML when compared to molecular dynamics (MD) simulations, which in turn are extremely intensive.12–17 To combine the benefits and limit the individual weaknesses of MD and ML, hybrid ML-MD approaches are now pursued, with the goal of accelerating simulations and improving the understanding of complex biomolecular systems (e.g. flexible molecular force fields, where ML tools are used to accelerate the simulation process).18–20 However, these tools are still at their infancy and their applications have to be fully explored.
Here, we developed a new computational design protocol to discover oligopeptides that self-assemble in nanostructured biomaterials by combining an unbiased AA selection (i.e. agnostic to chemical features) with coarse grain (CG) MARTINI forcefield (highly parameterized for natural amino acids), which yields a speed-up of 2–3 orders of magnitude compared to atomistic forcefield. We focused on tetrapeptides as n = 4 represents a wide but approachable sequences space (204 possible unique sequences) to screen and test computational unbiased methods, while possessing an amphiphilic form and the proven ability to self-assemble into nanofibrillar structures.21 This method allowed us to simulate the assembly of all the possible tetrapeptides without bias, resulting in the screening of appropriate side chain combinations to embed responsiveness to environmental stimuli, such as pH. As an example, the imidazole group of histidine has a pKa of 6.0, which allows controlling the AA protonated or deprotonated states across physiological pH. pH-induced control of imidazole's electrostatic interactions can be coupled with hydrophobic dissipative forces of aromatic amino acids, to provide pH control over tetrapeptide self-assembly behavior. This pH-triggered assembly of tetrapeptides can be used to engineer new biomaterials for drug delivery that assemble/disassemble in response to pH changes, nanofibrillar matrices for separation of large biomolecules like proteins, antimicrobial surfaces, and scaffolds to support cell growth. The protocol starts by generating the CG models of tetrapeptides that result from the permutation of 16 natural amino acids in the four positions, excluding C, M, W, and P. These four AA are excluded as they rarely appear in known naturally occurring oligopeptides that self-assemble. To provide pH-responsiveness for the transition between soluble to assembled peptides, we impose that each sequence must present at least one histidine residue to exploit the change in protonation state close to physiological pH. In total, ∼12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tetrapeptides with different sequences are screened. Each peptide is modelled twice, either with protonated or deprotonated histidine residue(s), leading to a total of ∼24000 different systems. For each peptide sequence, we model the aggregation propensity following a protocol described in previous works.22,23 Briefly, for each peptide sequence, we generate a periodic cubic box of 10 nm by side containing a random distribution of 60 peptides. The box is then solvated with MARTINI water beads, and counterions (Na+ and Cl−) are inserted to neutralize the system (Fig. 1). The final model consists of ∼7000 beads. For each peptide sequence, we generate three replicas of the box, leading to ∼72000 different models. The molecular systems are simulated with GROMACS 2021.4 package following protocols used in previous studies.24–26 Analyses of the simulations are performed using GROMACS tools and in-house tcl-scripts in VMD.27,28
000 tetrapeptides with different sequences are screened. Each peptide is modelled twice, either with protonated or deprotonated histidine residue(s), leading to a total of ∼24000 different systems. For each peptide sequence, we model the aggregation propensity following a protocol described in previous works.22,23 Briefly, for each peptide sequence, we generate a periodic cubic box of 10 nm by side containing a random distribution of 60 peptides. The box is then solvated with MARTINI water beads, and counterions (Na+ and Cl−) are inserted to neutralize the system (Fig. 1). The final model consists of ∼7000 beads. For each peptide sequence, we generate three replicas of the box, leading to ∼72000 different models. The molecular systems are simulated with GROMACS 2021.4 package following protocols used in previous studies.24–26 Analyses of the simulations are performed using GROMACS tools and in-house tcl-scripts in VMD.27,28
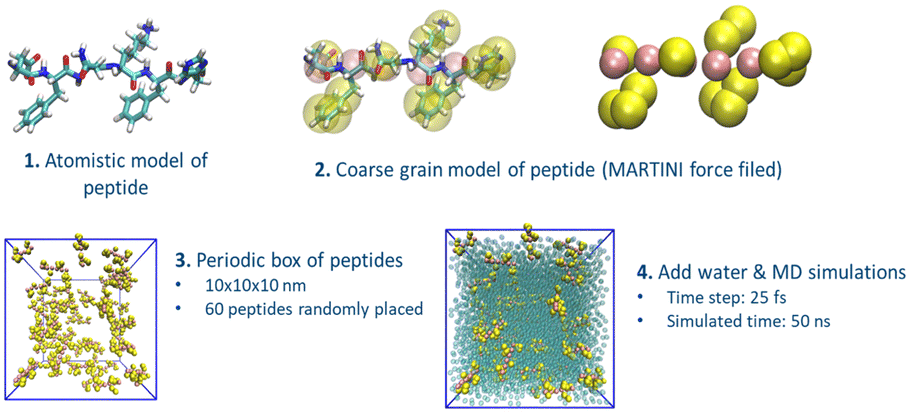 | ||
| Fig. 1 Schematics of the computational design protocol for peptide FNKF Starting from the atomistic model of the peptide (1), we built the coarse-grain (CG) representation (2). An ensemble of 60 CG homologues peptides is then places in a periodic box and fully solvated with explicit water. Each system is finally simulated for 50 ns. | ||
After the MD simulations (refer to ESI†), we ranked the peptides based on their aggregation potential, using a score similar to the one proposed by Frederix et al. with some modifications (Fig. 2).23 The scoring system aims to identify peptides that are prone to aggregate in large fibril-like clusters. The peptide score (total score – S) is obtained from:
| S = w1 × C + w2 × AP + w3 × SF + w4 × H | (1) |
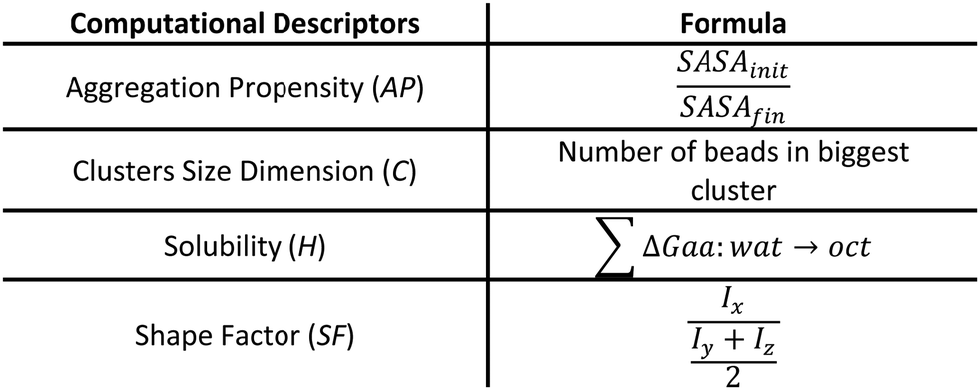 | ||
| Fig. 2 Descriptors and their mathematical definition. | ||
 | ||
| Fig. 3 (A) Total score (S) used to select peptides candidates. FFHH and FHYH were chosen from the top of the ranking, negative controls were picked from the middle rank (0.45 < S < 0.55, red cross). (B) Chemical structure of three tetrapeptides considered. (C) Initial (t = 0 ns) and final (t = 50 ns) different conformations for fibrillar-like peptides (upper panel) and beads-like (Video S2, ESI†) tetrapeptides. All the simulations start from a random distribution of the 60 homologues tetrapeptides in the water box, after 50 ns the promising sequences (e.g. HHFF, Video S1, ESI†) assume a 1D fibrillar conformation, while the negative controls (e.g. DHHR, Video S2, ESI†) are homogeneously distributed in the water box or have spherical conformation. | ||
Each term of the scoring function is normalized from 0 to 1 over its distribution using min–max normalization formula; the overall score S ranges from 0 to 1, where a score near 1 indicates a sequence that is likely to form large fibril-like aggregate and presents a suitable solubility. The C term represents the size of the largest peptide cluster observed at the end of the simulation (in terms of the number of beads). The size of the cluster is obtained through the clustsize tool of Gromacs. The larger the clusters formed the higher the propensity of the peptides to aggregate. The AP term is a measure of the aggregation propensity:
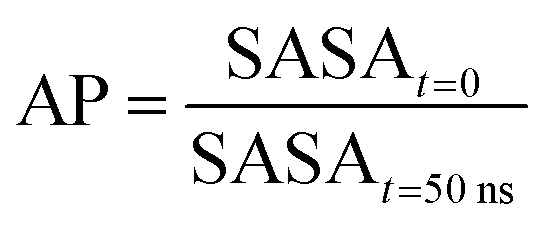 | (2) |
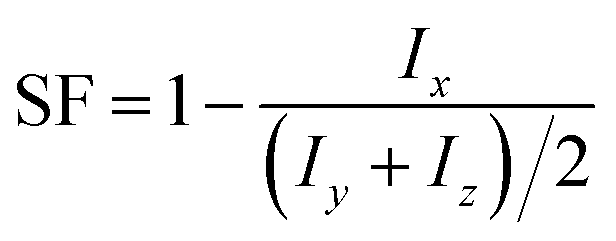 | (3) |
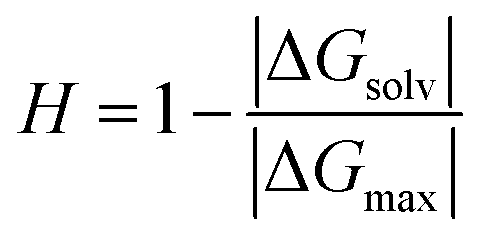 | (4) |
A high S-score with either the protonated or deprotonated form (indicating the propensity to form fibril-like aggregates, Fig. 3(C), Fig. S1 and Supporting Data File 1, ESI†) and a concurrent large value for the difference between the S-score in the protonated and deprotonated form (ΔS = |Sdeprotonated − Sprotonated|) were used as criteria to select the peptides to test experimentally. The latter condition favors peptides that upon the change of the protonation state can transition from assembled to disassembled states.
The computational screening provides the basis for a selection of the most promising peptides in term of self-assembling propensity (Fig. 3(B)). We first selected the peptide with the highest overall score at neutral pH (i.e., with deprotonated histidine residues). The best sequence is HHFF with an overall score of 0.846 (where C = 1.000, AP = 0.830, SF = 0.965, H = 0.503). This peptide has the largest cluster among all peptide sequences, a high aggregation propensity score, a shape factor close to 1 (indicating the propensity to form fibril-like aggregates) and is mildly hydrophobic. Generally, HHFF is expected to self-assemble in neutral-basic pH forming fibril-like aggregates.
A second peptide was chosen based on the highest difference between the score of the deprotonated form and the score of the protonated form. In this way, we aimed to identify a peptide that can self-assemble at basic pH, with deprotonated histidine residues and, at the same time, that is able to disaggregate at acidic pH, when histidine residues are protonated. The candidate peptide sequence for this class is FHYH, which showed a ΔS = 0.476, due to a Sdeprotonated = 0.826 (indicating a high tendency to self-assemble) and a Sprotonated = 0.350 (indicating a low tendency to self-assemble).
Finally, we chose sequences from the center of our ranking as negative controls, avoiding the bottom range of our ranking due to too severe and well-known hydrophilic behavior. All selected sequences, in the deprotonated form, present the highest score as defined in the work of Frederix et al.23 Their score only considers the aggregation propensity (AP) and the solubility of the sequence (H). Based on this scoring system, the selected candidate peptide sequences are DHHR, and VDKH. To validate our modeling, tetrapeptide DHHR, VDHK, FHYH, and HHFF with N-terminal acetylation and C-terminal amidation (purity >99%) were synthesized and tested in both protonated and deprotonated forms in phosphate buffer solutions. We found that DHHR and VDHK don’t show any gelation or precipitation at pH 5–9 (Fig. 4(A) and Fig. S3A, ESI†). DHHR generated micro-flakes (Fig. 4(D)) while VDHK generated a smooth film-like structure during the slow water evaporation process (Fig. S3B, ESI†). At an acidic pH of 5, FHYH and HHFF don’t assemble (Fig. 4(A)) and keep dispersing in solution with fiber-like structures when dried (Fig. 4(E) and (F), respectively). FHYH and HHFF, however, assemble at pH 7 and 9 (Fig. 4(B) and (C)), which corroborates our computational results. FHYH assembly generates transparent gel at a neutral or basic pH, which adheres to the bottom of the vial when turned upside down, whereas HHFF assembly results in the formation of nanofibrillar white precipitates in the vials that do not support the formation of a hydrogel. (Fig. 4(A)). ATR-FTIR was used to determine the secondary structure of assembled tetrapeptides by analyzing their spectra in the 1750–650 cm−1 region (Fig. S4A, ESI†). The Amide I resonance peaks at 1705–1595 cm−1 for DHHR, FHYH, and HHFF are depicted in Fig. S4B–D (ESI†), respectively. The broad Amide I resonance of DHHR and VDHK indicates a disordered structural conformation of the tetrapeptide (Fig. S3C and S4B, ESI†). These data corroborate with the lack of assembly in hydrogels (Fig. 4(A)) together with the formation of sparse nanoaggregates during solvent casting (Fig. 4(D)) and provide an experimental validation that a low (<0.6) S-score can predict a low tendency to assemble in ordered structures. Amide I peaks of FHYH and HHFF (Fig. S4C and D, ESI†), were centered on the 1635–1630 cm−1 region and attributed to β-sheet structures, indicating a disorder to order transition during assembly.29,30 Additionally, when pH increases to neutral or basic values and tetrapeptides become deprotonated, the contribution of ordered conformations in the Amide I peak becomes more prominent, denoting that higher-order assembly accrues with an increase in pH. These data corroborate the formation of hydrogels and SEM analysis, validating the use of a high S score (>0.8) to predict formation of ordered, assembled structures.
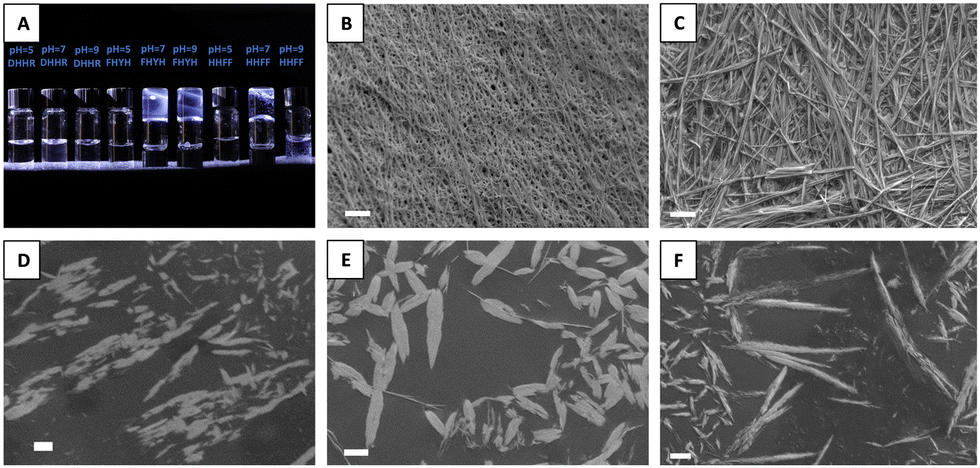 | ||
| Fig. 4 (A) Tetrapeptides in 50 mM phosphate buffer solutions. Scanning Electron Microscope (SEM) images of freeze-dried gels (B) FHYH and (C) HHFF post tetrapeptide assembly in 50 mM phosphate buffer solution at pH = 9. SEM images of (D) DHHR, (E) FHYH, and (F) HHFF nanoassemblies generated in water solution. Scale bar = 1 μm. | ||
In conclusion, by computationally screening and ranking all possible combinations of histidine-bearing tetrapeptides, we successfully identified peptides with pH-tunable gelation properties. Amino acids can change their protonation state based on local effects during self-assembly, as described by Tang et al.31 Considering intermediate protonation scenarios or implementing a constant pH molecular dynamics method, as described by the Tuttle group,32 could further improve the accuracy of the molecular models and could provide valuable insights into the behavior of the identified systems of interest. However, these approaches would require a significant increment in the computational cost, which would impair high-throughput in silico screening of different peptide sequences. In future works we can envision a first large-scale screening with fixed protonation, followed by a second-more accurate-filter with constant pH MD to select self-assembling peptide sequences. Tunable biomaterials with a pH-switch can find applications in releasing bioactive cargos molecules at specific tissue targets such as inflammatory sites, mineralizing bone, and tumors. Additionally, hydrogels that assemble in response to pH can be easily deployed in remote tissues by injection.
Conceptualization, YH, FR, AG, BM; methodology, YH, FR, AG; formal analysis, YH, FR; investigation, YH, FR; resources, AG and BM; data curation, YH, FR; writing, review and editing, YH, FR, AG, BM; supervision, AG, BM; funding acquisition, AG, BM. All authors have read and agreed to the published version of the manuscript.
BM acknowledges NSF (CMMI-1752172), ONR (N000142112402 and N000141912317), and MIT-IBM Watson AI Lab. AG acknowledges the CINECA award under the ISCRA initiative (grant code IsB26_W2EB and IsCa8_ReFPOX).
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. Tamamis, L. Adler-Abramovich, M. Reches, K. Marshall, P. Sikorski, L. Serpell, E. Gazit and G. Archontis, Biophys. J., 2009, 96, 5020 CrossRef CAS PubMed.
- G. F. Chen, T. H. Xu, Y. Yan, Y. R. Zhou, Y. Jiang, K. Melcher and H. E. Xu, Acta Pharmacol. Sin., 2017, 38, 1205 CrossRef CAS PubMed.
- K. A. Murray, C. J. Hu, S. L. Griner, H. Pan, J. T. Bowler, R. Abskharon, G. M. Rosenberg, X. Cheng, P. M. Seidler and D. S. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2206240119 CrossRef CAS PubMed.
- S. Y. Ow and D. E. Dunstan, Protein Sci., 2014, 23, 1315 CrossRef CAS PubMed.
- F. Sheehan, D. Sementa, A. Jain, M. Kumar, M. Tayarani-Najjaran, D. Kroiss and R. V. Ulijn, Chem. Rev., 2021, 121, 13869–13914 CrossRef CAS PubMed.
- N. Stephanopoulos, Chem, 2020, 6, 364–405 CAS.
- J. B. Matson, R. H. Zha and S. I. Stupp, Curr. Opin. Solid State Mater. Sci., 2011, 15, 225–235 CrossRef CAS PubMed.
- N. Habibi, N. Kamaly, A. Memic and H. Shafiee, Nano Today, 2016, 11, 41 CrossRef CAS PubMed.
- Y. Lin and C. Mao, Front. Mater. Sci., 2011, 5, 247–265 CrossRef PubMed.
- L. Jiang, D. Xu, T. J. Sellati and H. Dong, Nanoscale, 2015, 7, 19160–19169 RSC.
- T. A. Enache, A. M. Chiorcea-Paquim and A. M. Oliveira-Brett, Anal. Chem., 2018, 90, 2285–2292 CrossRef CAS PubMed.
- S. Doerr, M. Majewski, A. Pérez, A. Krämer, C. Clementi, F. Noe, T. Giorgino and G. De Fabritiis, J. Chem. Theory Comput., 2021, 17, 2355–2363 CrossRef CAS PubMed.
- Y. Liu, Y. H. Zhu, X. Song, J. Song and D. J. Yu, Briefings Bioinf., 2021, 22, 1–14 CrossRef PubMed.
- G. B. Goh, N. O. Hodas and A. Vishnu, J. Comput. Chem., 2017, 38, 1291–1307 CrossRef CAS PubMed.
- M. S. Friedrichs, P. Eastman, V. Vaidyanathan, M. Houston, S. Legrand, A. L. Beberg, D. L. Ensign, C. M. Bruns and V. S. Pande, J. Comput. Chem., 2009, 30, 864 CrossRef CAS PubMed.
- S. A. Bray, T. Senapathi, C. B. Barnett and B. A. Grüning, J. Cheminf., 2020, 12, 1–13 Search PubMed.
- Z. Qin, L. Wu, H. Sun, S. Huo, T. Ma, E. Lim, P. Y. Chen, B. Marelli and M. J. Buehler, Extreme Mech. Lett., 2020, 36, 100652 CrossRef.
- S. Chmiela, H. E. Sauceda, K.-R. Müller and A. Tkatchenko, Nat. Commun., 2018, 9, 3887 CrossRef PubMed.
- G. G. Scott, T. Börner, M. E. Leser, T. J. Wooster and T. Tuttle, Front. Chem., 2022, 10, 822868 CrossRef CAS PubMed.
- A. van Teijlingen and T. Tuttle, J. Chem. Theory Comput., 2021, 17, 3221–3232 CrossRef CAS PubMed.
- S. Alshehri, H. H. Susapto and C. A. E. Hauser, Biomacromolecules, 2021, 22, 2094–2106 CrossRef CAS PubMed.
- P. W. J. M. Frederix, R. V. Ulijn, N. T. Hunt and T. Tuttle, J. Phys. Chem. Lett., 2011, 2, 2380–2384 CrossRef CAS PubMed.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed.
- N. Bono, B. Coloma Smith, F. Moreschi, A. Redaelli, A. Gautieri and G. Candiani, Nanoscale, 2021, 13, 8333–8342 RSC.
- A. Gautieri, S. Vesentini, A. Redaelli and M. J. Buehler, Int. J. Mater. Res., 2009, 100, 921–925 CrossRef CAS.
- A. Gautieri, M. Beeg, M. Gobbi, F. Rigoldi, L. Colombo and M. Salmona, Int. J. Mol. Sci., 2019, 20, 4641 CrossRef CAS PubMed.
- S. J. Marrink, H. Jelger Risselada, S. Yefimov, D. Peter Tieleman and A. H. de Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- X. Hu, D. Kaplan and P. Cebe, Macromolecules, 2006, 39, 6161–6170 CrossRef CAS.
- D. E. Clarke, C. D. J. Parmenter, A. Oren Scherman, D. E. Larke, O. A. Scherman and D. D. Parmenter, Angew. Chem., Int. Ed., 2018, 57, 7709–7713 CrossRef CAS PubMed.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS PubMed.
- A. Van Teijlingen, H. W. A. Swanson, K. H. A. Lau and T. Tuttle, J. Phys. Chem. Lett., 2022, 13, 4046–4051 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc02412a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |