
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A supramolecular helicate with two independent Fe(II) switchable centres and a [Fe(anilate)3]3− guest†
Leoní A.
Barrios
 *ab,
Simon. J.
Teat
c,
Olivier
Roubeau
*de and
Guillem
Aromí
*ab
*ab,
Simon. J.
Teat
c,
Olivier
Roubeau
*de and
Guillem
Aromí
*ab
aDepartament de Química Inorgànica i Orgànica, Secció Química Inorgànica, Universitat de Barcelona, Barcelona, Spain
bInstitute of Nanoscience and Nanotechnology of the University of Barcelona (IN2UB), Barcelona, Spain. E-mail: leoni.barrios@ub.edu; aromi@ub.edu
cAdvanced Light Source, Berkeley Laboratory, 1 Cyclotron Road, Berkeley, California 94720, USA
dInstituto de Nanociencia y Materiales de Aragón (INMA), CSIC-Universidad de Zaragoza, Zaragoza, Spain. E-mail: roubeau@unizar.es
eDepartamento de Física de la Materia Condensada, Universidad de Zaragoza, Zaragoza, Spain
First published on 5th August 2023
Abstract
A biphenyl-spaced bis-pyrazolylpyridine ligand interacts with ferrous ions to engender a dimetallic helical coordination cage that encapsulates an Fe3+ tris-anilate complex. The host–guest interaction breaks the symmetry of the Fe2+ centers causing a differential spin crossover behavior in them that can be followed in great detail crystallographically.
Coordination supramolecular chemistry, where metals and ligands are programmed to assemble into structures with predetermined geometries, is a rich source of multifunctional molecular materials.1–3 In this context, the incorporation of active guest species inside the cavities of functional coordination cages represents a very convenient avenue to achieve synergies by combining properties.4–7 An interesting case of host functionality results from the presence of Fe2+ centres with spin crossover (SCO) switching properties.8–14 Of the latter, most of the reported instances involve tetrahedral Fe4 or cubic Fe8 cages.10,12,13,15,16 However, much simpler hosts involving only two iron centers are rarely found encapsulating any species. For some time, we have been exploring host–guest synergies in dinuclear Fe2+ triple stranded supramolecular helicates ([Fe2L3]4+) made of bis-pyrazolylpyridine ligands with various spacers. Thus, a 40 K shift to the SCO of a [Fe2(L1)3]4+ host was observed by simply swapping a Cl− by a Br− guest, both in the solid state and in solution (spacer of L1, phenylene).17,18 Furthermore, the symmetry and size of the guest SiF62− made it a template for selectively assembling two different ligands into a heteroleptic [Fe2(L1)(L2)2]4+ helicate19 (spacer of L2, biphenyl; Fig. 1). Otherwise, three L2 ligands engender a [Fe2(L2)3]4+ scaffold with a larger internal void perfectly suited to encapsulate a [M(ox)3]3− coordination complex (ox2− = oxalate; M = Cr3+, Fe3+). It was found that the influence of the host enhances dramatically the spin–lattice relaxation time of the guest magnetic moment (Cr3+)20 or supresses its quantum coherence (Fe3+).21 Incorporation of these complexes was enabled because the cavity of the host exhibits three wide windows with openings pointing to directions 120° from each other and perpendicular to the molecular axis. Therefore, octahedral coordination complexes with extended ligands may fit inside the [Fe2(L2)3]4+ functional host, allowing to incorporate a myriad of functional properties. In this respect, complexes of the type [M(ani)3]n− (M = various metals; ani = anilate derivatives) are an appealing choice as guest, since they are part of a prolific source of unique properties linked to electron transfer phenomena.22–25
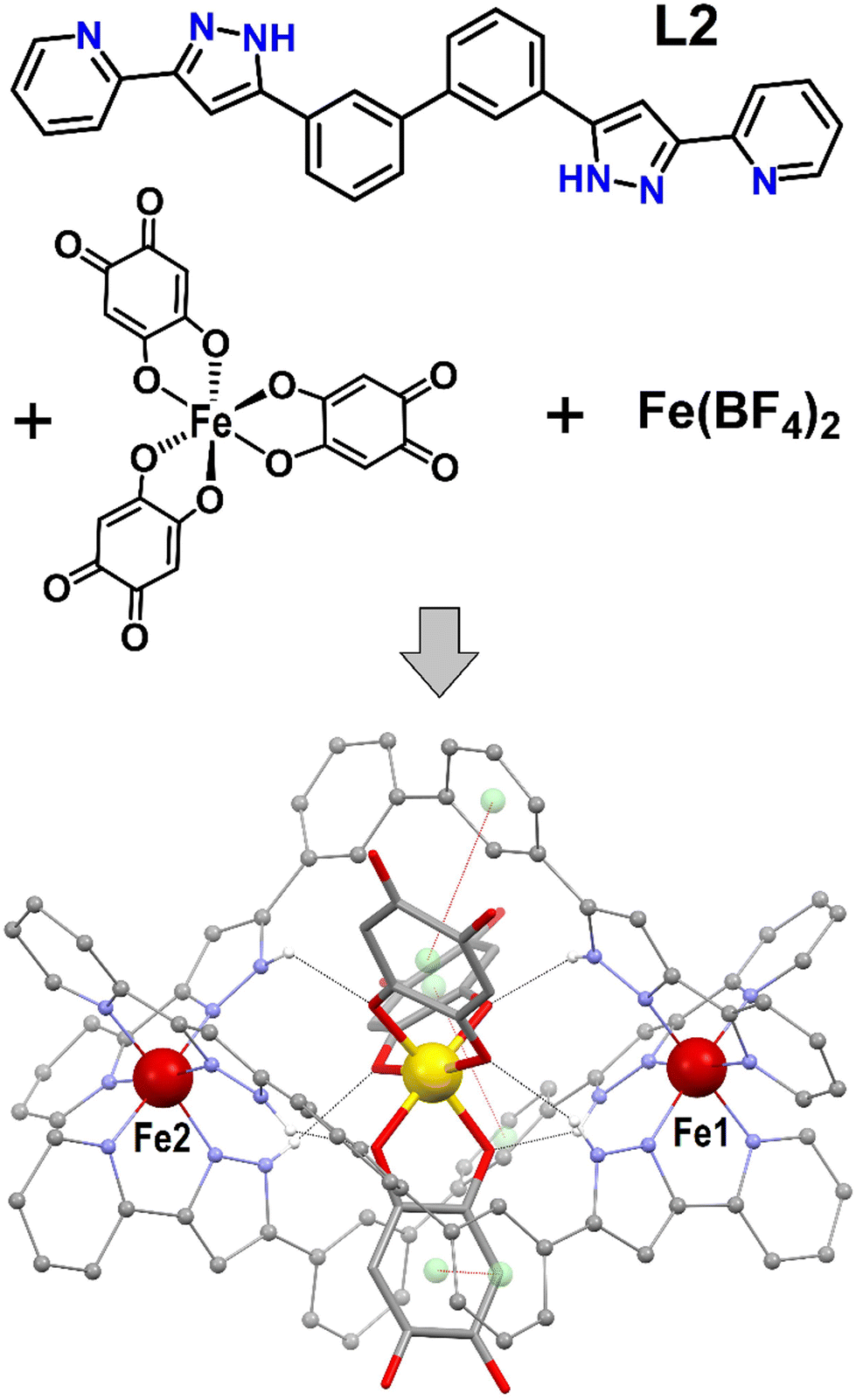 | ||
| Fig. 1 (top) Reaction scheme to produce 1, showing the molecular structure of ligand L2 (3,3′-bis(3-(pyridin-2-yl)-1H-pyrazol-5-yl)-1,1′-biphenyl) and of the complex cation [Fe(anilate)3]3−. (bottom) Representation of the supramolecular assembly ([Fe(anilate)3]@[Fe2(L2)3])+ of 1. Large red balls are Fe(II), the yellow ball is Fe(III), remaining red is O, grey is C, purple is N and white is H (only hydrogen from N–H groups shown). Black dashed lines are H-bonds. Red dashed lines are supramolecular π⋯π interactions between the anilate and aromatic rings of the host, where centroids are shown as green balls. | ||
The presence of [Fe(anilate)3]3− in a solution of Fe(BF4)2 and L2 (Fig. 1) in dry methanol under inert atmosphere, generates crystals after two weeks, of the host–guest supramolecular assembly [Fe(anilate)3]@[Fe2(L2)3](BF4) (1). Crystals of 1 are very robust and amenable to variable temperature single crystal X-ray diffraction (SCXRD) determinations (Tables S1–S3, ESI†). When measured fresh, they exhibit a cubic lattice from the Sohncke space group P213, with three lattice solvent molecules of MeOH per unit of 1, and four such units per cell. The asymmetric unit contains 1/3 of 1, with three Fe centers in special positions affected by three fold symmetry, 1/3 of a BF4− anion disordered by symmetry, and one crystallographically unique ligand L2, together with one MeOH molecule. The main supramolecular construct (Fig. 1 and Fig. S1–S3, ESI†) consists of a pseudo-octahedral [Fe(anilate)3]3− coordination complex positioned inside the cavity of a triple stranded supramolecular host [Fe2(L2)3]4+. The latter is made of two Fe2+ centres, linked to each other by three L2 ditopic ligands. At both ends of a biphenyl spacer, each ligand exhibits a bis-pyrazolylpyridine chelating moiety that coordinates one metal. When coordinated, L2 still features three C–C links with free rotation, providing the needed flexibility to accommodate a helical arrangement around a guest-adapted cavity.
The overall twist of the helicate can be gauged by the torsion angle Npy–Fe–Fe–Npy (in moving from one end to the other of the helicate on the same ligand), which measures 179.3° at 100 K. This pronounced rotation is ensured by the twist generated at both octahedral metal sites, the C–C bond between the pyrazolylpyridine and the biphenyl (two torsion angles of 2.41°) and the C–C bond within the biphenyl (torsion of 49.54°). At 100 K, the average Fe–N bond distances are 1.97(3) (Fe1) and 1.94(6) Å (Fe2), consistent with both metal centers being in the low spin (LS) state.26 The guest exhibits very similar metric parameters (Table S4, ESI†) as it does within the salt [(Ph)4P]3[Fe(anilate)3],27 used as starting material to obtain 1. It is an octahedral ferric complex of three double deprotonated 3,6-dihydroxy-1,4-benzoquinone (DHBQ) units. This complex is located inside the large open cavity offered by the [Fe2(L2)3]4+ helicate, which exhibits trigonal symmetry and three key features. First, six N–H moieties with approximately trigonal antiprismatic symmetry, poised to form [N–H⋯O] hydrogen bonds with the six oxygen donor atoms of the [Fe(anilate)3]3− complex. Second, three large windows in the appropriate disposition to allow the extended anilate moieties traversing the cage, thus enabling the encapsulation of this large species. Third, three aromatic rings from the biphenyl spacers forming π⋯π stacking interactions with the anilate ligands, contributing to the host–guest recognition (inter-centroid distances of 3.812 Å). The three interactions occur with biphenyl fragments always on the same side of the helicate (Fig. 1 and Fig. S3, ESI†), causing the Fe2+ ion of this side, Fe1, to lye slightly closer to the central Fe3+ than the other ferrous center, Fe2 (5.256 vs. 5.357 Å). The crystal lattice only contains one of the two possible enantiomers of 1. In the structure described here (Fig. 1), the rotation of the helical host evolves clockwise from the end closer to the observer towards the other end as does the rotation of the central octahedral guest. The racemic nature of the whole system is ensured by the presence of the corresponding enantiomorphic crystals, as verified following multiple structural determinations. The composite assemblies are held together through a dense network of weak van der Waals interactions, none standing out as significantly stronger. The thermal evolution of the structural parameters was studied in detail by SCXRD. This study was hampered by anomalies ascribed to the desorption of MeOH molecules of the lattice upon heating (see ESI† for full crystallographic and magneto-thermal study). This drawback was circumvented by studying the equivalent system in the absence of the lattice solvent. Fresh crystals of 1 desolvated upon standing in grease at room temperature, exhibit the same structural features as described above (Table S3, ESI†) but no lattice molecules. Specifically, the Fe–N bond distances at 100 K (average of 1.976(11) and 1.978(11) Å for Fe1 and Fe2, respectively) correspond again to these for the metals in the LS state. The structure of the solvent-depleted version of 1 was determined at various temperatures upon heating and cooling. The good quality of the data persisted during the whole thermal cycle with no anomalies, allowing to examine the evolution of all the metric parameters with temperature. The crystal lattice parameter a shows a linear dependence with the temperature (Fig. 2), consistent with thermal expansion and therefore not affected by the process of SCO at the ferrous sites (see below). The molecular parameters show almost no variations except for the average Fe–N distances, which increase with heating in both Fe centres as predicted for gradual transitions from the low to the high-spin (HS) state (Fig. 2). Interestingly, the transitions do not take place at the same temperature; for Fe1 it begins around 300 K whereas for Fe2 it starts already below 250 K. The observed superposition of the data as obtained upon warming and cooling indicates that the SCO process is reversible. Differential SCO behaviour of the Fe2+ centres in dinuclear molecules has been investigated for some time,28,29 with an early example of a [Fe2L3]4+ helicate displaying a stable [LS–HS] mixed-state reported more than a decade ago.30 We have later described in detail site selective (or ordered) [LS–LS] → [LS–HS]20 or [LS–HS] → [HS–HS]17 SCO in helicates, following structurally the transition of the affected Fe atoms through variable temperature SCXRD.
 | ||
| Fig. 2 Thermal evolution of the cell parameter a (black circles), average Fe–N bond distances (grey symbols) and N⋯O separations of the pyrazole-anilate hydrogen bonds (empty symbols) of desolvated 1. Lines are a linear fit in the case of the cell parameter a, and otherwise only a guide to the eye. | ||
We are not aware of a well-characterized case of simultaneous SCO, occurring at different temperatures by two different metal atoms in one molecule. The likely cause of the differential magnetic behavior of Fe1 vs. Fe2 is the asymmetry caused to the host by the [Fe(anilate)3]3− guest; the three π⋯π interactions of the anilate rings with the host occur on the same side of the later (Fig. 1 and Fig. S3, ESI†). This may restrict slightly the structural changes required for the LS → HS SCO. Additionally, these contacts cause a slight shortening of the pyrazole-anilate [N–H⋯O] hydrogen bonding interactions near Fe1, likely causing an increase of the ligand field around this ion. Both effects are expected to slightly stabilize the LS state of Fe1 compared to Fe2, as observed crystallographically. The effect of the hydrogen bond on the SCO for related coordination spheres around Fe(II) has been studied.31 Interestingly, in compound 1, both types of N⋯O separations also depict differential variations associated the respective SCO processes (Fig. 2 and Table S3, ESI†), while their difference is maintained throughout the whole thermal cycle.
The SCO processes unveiled by SCXRD were studied through variable temperature magnetic susceptibility. Measurements were performed on a polycrystalline sample depleted of solvate MeOH molecules by keeping it in open atmosphere for several hours. At low temperature, the χT value (χ is the molar paramagnetic susceptibility) exhibits a wide plateau at ca. 5.03 cm3 Kmol−1 corresponding to the Fe3+ centre of the [Fe(anilate)3]3− guest being in the HS state S = 5/2 (Fig. 3). This is consistent with the decline of χT below 20 K, expected from the zero-field splitting or Zeeman effects for such spin system in this temperature regime. The isothermal magnetization data at 5 K indeed agrees well with the Brillouin function for S = 5/2 and g = 2.06 (Fig. 3, inset). The χT vs. T curve obtained when warming from 4 to 390 K shows a gradual increase from the plateau for T above 150 K consistent with the LS to HS transition of the Fe2+ centres of 1 as observed by SCXRD (see above). Since both transitions are very gradual, they cannot be discriminated by the bulk magnetization curve, which features the added response at any given temperature. Upon subsequent cooling and further thermal cycles, the χT vs. T plot shows a similar reproducible spin transition, delineating a slightly different path than seen for the initial warming branch (Fig. S8, ESI†). This is ascribed to the fact that the warming is likely accompanied by desorption of H2O lattice molecules that have entered the lattice prior to the experiment (as corroborated by microanalysis, ESI†), which is very common for molecular SCO systems.30,32 The broad nature of the spin switching is consistent with the absence of strong intermolecular interactions. Therefore, the pace of the temperature cycle is not expected to affect the kinetics of the transition. This was corroborated for two different temperature scan rates, which led to superimposable χT vs. T curves (Fig. S9, ESI†). Altogether, the bulk magnetization results are in full agreement with the magnetic processes revealed by the crystallographic results.
 | ||
| Fig. 3 Thermal evolution upon heating of the χT product of desolvated 1, χ being the molar paramagnetic susceptibility. Inset: Field dependence of the magnetization at 5 K. The red line is the Brillouin function for S = 5/2 and g = 2.06 at this temperature. | ||
We have shown the design of a ligand, L2, with the structural and electronic properties conducive to a [Fe2(L2)3]4+ helicate with SCO properties capable to encapsulate extended coordination complexes. The interaction of the guest with the host generates a symmetry break causing a differential magnetic behaviour of the Fe2+ centers of the cage. The distinct switching properties can be followed in detail through variable temperature SCXRD. The ability to encapsulate coordination complexes with an octahedral MO6 coordination geometry and extended ligands opens a wide range of possibilities to study the synergy of different properties within such type of supramolecular assemblies. These properties include single ion slow relaxation of the magnetization, quantum coherence, photoluminescence or electron transfer. It also opens an avenue to explore potential metal organic frameworks of host/guest systems, where the guests would act also as connecting nodes.
This research was supported by the Spanish Ministry of Science (TED2021-129214B-I00, PID2020-118329RB-I00, PRTR-C17.I1), the Aragón government (E31_20R PLATON) and the EU (FET-OPEN grant 862893 FATMOLS and Next Generation EU), and used resources of the ALBA synchrotron and of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DEAC02-05CH11231. G. A. thanks the Generalitat de Catalunya for Next Generation co-fund (PRTR-C17.I1) and the prize ICREA Academia 2018.
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. H. Dinolfo and J. T. Hupp, Chem. Mater., 2001, 13, 3113–3125 CrossRef CAS.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022–2043 CrossRef CAS PubMed.
- A. Schmidt, A. Casini and F. E. Kuhn, Coord. Chem. Rev., 2014, 275, 19–36 CrossRef CAS.
- M. Morimoto, S. M. Bierschenk, K. T. Xia, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Catal., 2020, 3, 969–984 CrossRef CAS.
- X. Jing, C. He, L. Zhao and C. Duan, Acc. Chem. Res., 2019, 52, 100–109 CrossRef CAS PubMed.
- A. Galan and P. Ballester, Chem. Soc. Rev., 2016, 45, 1720–1737 RSC.
- J. Zheng, L. K. S. Von Krbek, T. K. Ronson and J. R. Nitschke, Angew. Chem., Int. Ed., 2022, 61, e202212634 CAS.
- A. J. McConnell, Supramol. Chem., 2018, 30, 858–868 CrossRef CAS.
- I. C. Berdiell, T. Hochdörffer, C. Desplanches, R. Kulmaczewski, N. Shahid, J. A. Wolny, S. L. Warriner, O. Cespedes, V. Schünemann, G. Chastanet and M. A. Halcrow, J. Am. Chem. Soc., 2019, 141, 18759–18770 CrossRef CAS PubMed.
- L. Li, A. R. Craze, O. Mustonen, H. Zenno, J. J. Whittaker, S. Hayami, L. F. Lindoy, C. E. Marjo, J. K. Clegg, J. R. Aldrich-Wright and F. Li, Dalton Trans., 2019, 48, 9935–9938 RSC.
- A. Ferguson, M. A. Squire, D. Siretanu, D. Mitcov, C. Mathonière, R. Clérac and P. E. Kruger, Chem. Commun., 2013, 49, 1597 RSC.
- N. Struch, C. Bannwarth, T. K. Ronson, Y. Lorenz, B. Mienert, N. Wagner, M. Engeser, E. Bill, R. Puttreddy, K. Rissanen, J. Beck, S. Grimme, J. R. Nitschke and A. Lützen, Angew. Chem., Int. Ed., 2017, 56, 4930–4935 CrossRef CAS PubMed.
- R. W. Hogue, S. Singh and S. Brooker, Chem. Soc. Rev., 2018, 47, 7303–7338 RSC.
- R. A. Bilbeisi, S. Zarra, H. L. C. Feltham, G. N. L. Jameson, J. K. Clegg, S. Brooker and J. R. Nitschke, Chem. – Eur. J., 2013, 19, 8058–8062 CrossRef CAS PubMed.
- D.-H. Ren, D. Qiu, C.-Y. Pang, Z. Li and Z.-G. Gu, Chem. Commun., 2015, 51, 788–791 RSC.
- M. Darawsheh, L. A. Barrios, O. Roubeau, S. J. Teat and G. Aromí, Chem. – Eur. J., 2016, 22, 8635–8645 CrossRef CAS PubMed.
- D. Y. Aleshin, R. Diego, L. A. Barrios, Y. V. Nelyubina, G. Aromí and V. V. Novikov, Angew. Chem., Int. Ed., 2022, 61, e202110310 CAS.
- N. Capó, L. A. Barrios, J. Cardona, J. Ribas-Ariño, S. J. Teat, O. Roubeau and G. Aromí, Chem. Commun., 2022, 58, 10969–10972 RSC.
- M. Darawsheh, L. A. Barrios, O. Roubeau, S. J. Teat and G. Aromí, Angew. Chem., Int. Ed., 2018, 57, 13509–13513 CrossRef CAS PubMed.
- L. A. Barrios, R. Diego, M. Darawsheh, J. I. Martínez, O. Roubeau and G. Aromí, Chem. Commun., 2022, 58, 5375–5378 RSC.
- J. A. Degayner, K. Wang and T. D. Harris, J. Am. Chem. Soc., 2018, 140, 6550–6553 CrossRef CAS PubMed.
- P. Sadhukhan, S.-Q. Wu, J. I. Long, T. Nakanishi, S. Kanegawa, K. Gao, K. Yamamoto, H. Okajima, A. Sakamoto, M. L. Baker, T. Kroll, D. Sokaras, A. Okazawa, N. Kojima, Y. Shiota, K. Yoshizawa and O. Sato, Nat. Commun., 2021, 12, 4836 CrossRef CAS PubMed.
- J. Chen, Y. Sekine, A. Okazawa, H. Sato, W. Kosaka and H. Miyasaka, Chem. Sci., 2020, 11, 3610–3618 RSC.
- J. Chen, Y. Sekine, Y. Komatsumaru, S. Hayami and H. Miyasaka, Angew. Chem., 2018, 130, 12219–12223 CrossRef.
- G. A. Craig, O. Roubeau and G. Aromí, Coord. Chem. Rev., 2014, 269, 13–31 CrossRef CAS.
- M. Atzori, L. Marchiò, R. Clérac, A. Serpe, P. Deplano, N. Avarvari and M. L. Mercuri, Cryst. Growth Des., 2014, 14, 5938–5948 CrossRef CAS.
- A. B. Gaspar, M. C. Muñoz and J. A. Real, J. Mater. Chem., 2006, 16, 2522–2533 RSC.
- J. J. M. Amoore, C. J. Kepert, J. D. Cashion, B. Moubaraki, S. M. Neville and K. S. Murray, Chem. – Eur. J., 2006, 12, 8220–8227 CrossRef CAS PubMed.
- R. J. Archer, C. S. Hawes, G. N. L. Jameson, V. McKee, B. Moubaraki, N. F. Chilton, K. S. Murray, W. Schmitt and P. E. Kruger, Dalton Trans., 2011, 40, 12368–12373 RSC.
- G. Lemercier, N. Bréfuel, S. Shova, J. A. Wolny, F. Dahan, M. Verelst, H. Paulsen, A. X. Trautwein and J.-P. Tuchagues, Chem. – Eur. J., 2006, 12, 7421–7432 CrossRef CAS PubMed.
- C. Bartual-Murgui, C. Codina, O. Roubeau and G. Aromí, Chem. – Eur. J., 2016, 22, 12767–12776 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, crystallography, physical measurements. CCDC 2260940–2260952, 2261719–2261733, and 2261757–2261767. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc02278a |
| This journal is © The Royal Society of Chemistry 2023 |