
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVariable temperature kinetic isotope effects demonstrate extensive tunnelling in the C–H activation reactivity of a transition metal-oxo complex†
Joseph E.
Schneider
a,
McKenna K.
Goetz
 ab and
John S.
Anderson
*a
ab and
John S.
Anderson
*a
aDepartment of Chemistry, University of Chicago, Chicago, Illinois 60637, USA. E-mail: jsanderson@uchicago.edu
bDepartment of Chemistry, University of Wisconsin-Madison, Madison, Wisconsin 53706, USA
First published on 12th June 2023
Abstract
There has been recent interest about how the rates of concerted proton electron transfer (CPET) are affected by the thermodynamic parameters of intermediates from stepwise PT or ET reactions. Semiclassical arguments have been used to explain these trends despite the importance of quantum mechanical tunneling in CPET reactions. Here we report variable temperature kinetic isotope effect (KIE) data for the reactivity of a terminal Co-oxo complex with C–H bonds. The KIEs for the oxidation of both 9,10-dihydroanthracene (DHA) and fluorene have significant tunneling contributions and fluorene has a largely temperature-insensitive KIE which is inconsistent with semiclassical models. These findings support recent calls for a more detailed understanding of tunneling effects in thermodynamically imbalanced CPET reactions.
The activation of C–H bonds by transition metal-oxo complexes is a central reaction in biological and synthetic systems.1,2 It can be viewed as a subset of concerted proton electron transfer (CPET) reactions in which an electron in the C–H bond is transferred to the metal and the proton is transferred to an oxo ligand. The broad interest in transition metal-oxo reactivity, and CPET reactions more generally, has motivated a wide array of experimental and theoretical studies to better understand C–H activation by transition metal-oxo complexes.
Recently, there has been substantial discussion around asynchronous or imbalanced CPET where the thermodynamic energy of transferring either only an electron or a proton to generate formally stepwise electron transfer (ET) or proton transfer (PT) intermediates has a large influence on the rate of a concerted reaction (Scheme 1, top). There is building experimental evidence attesting to the importance of ET and PT free energies in CPET reactions,3–14 but the substantial quantum tunneling known to occur in CPET reactions has raised questions about whether or not the physical underpinnings of these trends is properly understood.15–17 Specifically, most free energy trends are couched in a semiclassical framework, whereas tunneling in CPET reactions is a fundamentally nonclassical quantum phenomenon.
 | ||
| Scheme 1 Top: Square scheme depicting imbalanced CPET. Bottom: Compounds and reactions discussed in this work. | ||
To better understand the intersection of proton tunneling and asynchronous CPET reactivity, we used variable temperature kinetic isotope effect (VT-KIE) studies to evaluate the nature of proton tunneling in the reactivity of a terminal Co-oxo complex which displays imbalanced reactivity with C–H bonds (Scheme 1, bottom).6 We find significant tunneling with 9,10-dihydroanthracene (DHA) and even more so with fluorene. These observations demonstrate that proton tunneling must be treated as an inherent feature, and not a correction to, physical models of imbalanced CPET reactivity. They further motivate theoretical studies aimed at reconciling quantum mechanical tunneling with more classical free energy relationships.
Reactivity trends in CPET, including imbalanced or asynchronous CPET, are often explained by evoking textbook structure-reactivity relationships, such as the influence of stepwise intermediates invoked in More-O’Ferrall–Jenks plots.18 Such analyses are based on a semiclassical view of the transferring proton, which is pictured in the transition state as localized midway across the reaction coordinate. Tunnelling effects can be added as a small correction in this semiclassical framework,19,20 but are fundamentally treated as a secondary perturbative effect. In contrast, modern theoretical treatments of CPET reactions stress the importance of treating the transferring proton as a quantum mechanical particle.21–23 In these theories, the entire H-atom directly tunnels from a vibronic state localized on the reactant to a vibronic state localized on the product. The proton is treated with a quantum mechanical wavefunction that is never localized halfway across the reaction coordinate. While this theory is firmly established, experimental investigations into the nature of proton tunneling are more limited. Variable temperature kinetic isotope effect (VT-KIE) experiments are the most rigorous way to evaluate tunneling reactivity and have revealed that tunneling is highly optimized in enzymatic reactivity.24,25 However, there have been comparably few studies on the temperature dependence of kinetic isotope effects in molecular systems, particularly with transition metal-oxo complexes.
VT-KIE studies inform on tunneling by breaking the KIE down into effects on the activation energy (Ea) and the Arrhenius prefactor (A).19,24,25 Analogous to rate constants, a plot of ln(KIE) against 1/RT is expected to be linear. The slope, Ea(D) − Ea(H), gives the isotope effect on Ea. The intercept, ln(AH/AD), gives the isotope effect on A. In semiclassical treatments KIEs are interpreted as the loss of zero-point energy in the transition state. This creates an isotope effect on Ea (slope), with minimal effect on A (intercept). Based on the frequencies of typical C–H and C–D bonds, this loss of zero-point energy can explain a value of Ea(D) − Ea(H) as high as ∼1.4 kcal mol−1, which corresponds to a KIE of ∼10 at room temperature. Proton tunneling is expected to increase the magnitude of kH and decrease its temperature dependence. This would result in larger KIEs and an extrapolated intercept of ln(AH/AD) much less than 0. Therefore, the traditional markers of substantial tunneling effects assayed via variable temperature KIE measurements have been observation of a KIE greater than 10, of a slope Ea(D) − Ea(H) greater than 1.4 kcal mol−1, or of an intercept ln(AH/AD) less than −0.7.19
The above scenario considers significant tunneling effects, but still relies on a semiclassical framework. However, if the transferring proton is treated as a quantum mechanical particle positioned in discrete vibrational energy states, then both H and D can tunnel extensively during a reaction. Without complicating factors such as variations in tunneling distance, this leads to a largely temperature independent KIE, and thus a small isotope effect on the slope, Ea(D) − Ea(H).23 Instead, the isotope effect will primarily manifest as a positive intercept, ln(AH/AD). This results in the counter-intuitive scenario where more extensive tunneling manifests in a smaller slope, generally smaller KIEs, and the above-mentioned positive intercept. A significantly positive intercept, above 0.4 as a conservative value, indicates this most extensive tunneling regime, even with traditionally small KIE values (<10). Notably, this limiting regime of extensive tunneling can only be observed through variable temperature KIE measurements.
With these experimental benchmarks in mind, we measured the temperature dependent KIEs of PhB(tBuIm)3CoIIIO (CoO) reacting with both fluorene and DHA (Scheme 1, Fig. 1, and Table 1). We measured KIEs for fluorene oxidation by competition, analyzing the deuteration of the dimerized radical product (bifluorenyl) from reactions with d1-fluorene (intramolecular KIEs) and with 50% d0-fluorene/50% d2-fluorene (intermolecular KIEs). This allowed us to determine both the primary and secondary KIEs for this reaction. In contrast, the KIEs from the reaction of CoO with DHA were too large for analysis via product composition; instead, we measured the KIEs by separately measuring kH and kD for d0-DHA and d4-DHA, respectively. Data analysis accounted for the true isotopic composition of the starting substrates (see ESI†). For completeness, we also performed kinetic measurements for reactivity of CoO with fluorene. Similar magnitudes were found for most of the KIE values; however, slight curvature in the variable temperature data arising from experimental artifacts made it difficult to confidently estimate the slope ln(AH/AD) (see ESI†). Thus, we discuss the values obtained from the competition experiments.
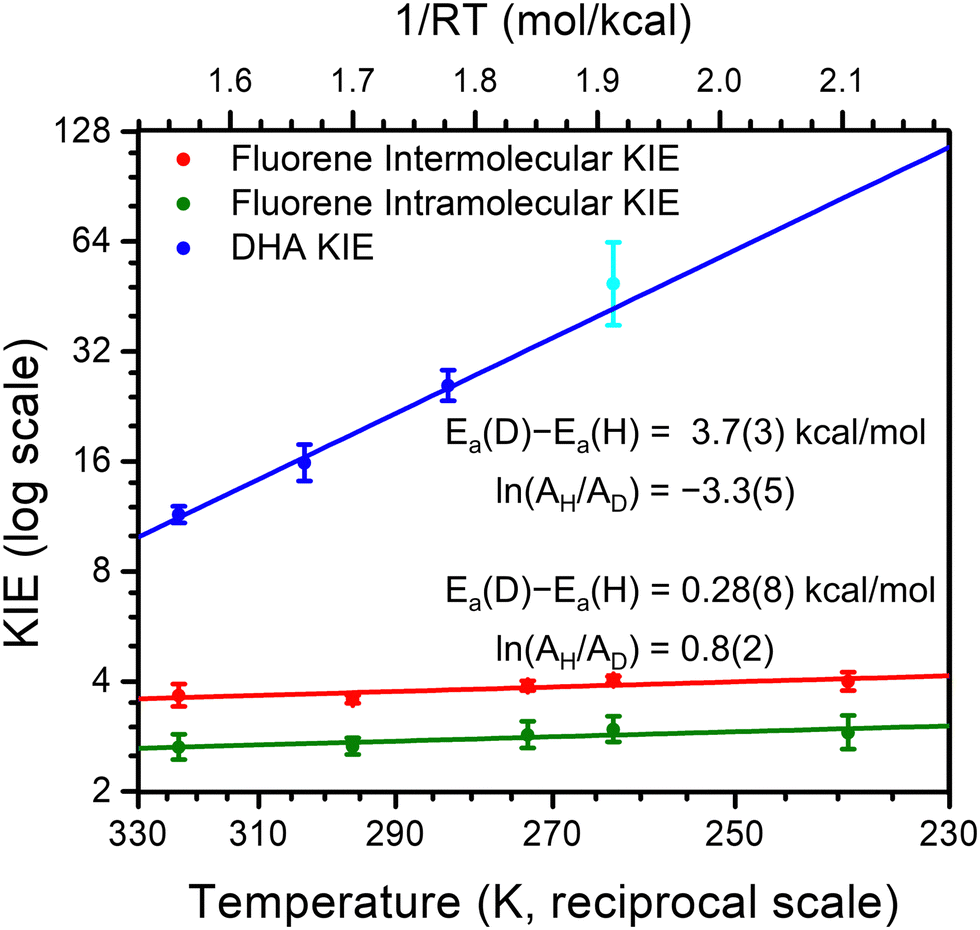 | ||
Fig. 1 Plots of ln(KIE) vs. 1/RT for the reactions of CoO with fluorene and DHA and the corresponding linear fits. The cyan colored data point at 263 K for DHA had kD determined via initial rates. This data point is graphed to demonstrate consistency with the other data points, but not included in the linear fits to variable temperature. The activation parameters shown for fluorene are for the primary KIE; the secondary isotope effect has Ea(D) − Ea(H) = 0.01(6), ln(AH/AD) = 0.1(1). For graphical purposes, the observed and predicted proton proportions (pH) and deuteron proportions (pD) from the competition data are displayed as KIE ≈ pH/pD for intermolecular competition between 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d0-fluorene d2-fluorene and as KIE ≈ pD/pH for intramolecular competition within d1-fluorene. :1 d0-fluorene d2-fluorene and as KIE ≈ pD/pH for intramolecular competition within d1-fluorene. | ||
| Reaction | Ref. | E a(D) − Ea(H) (kcal mol−1) | ln(AH/AD) |
|---|---|---|---|
| a Activation of a ligand C–H bond. b Two distinct sets of parameters are reported for this compound. c Values reported in DCM; similar results were also reported in MeCN. d Sigmatropic rearrangement to 4H-Quinolozine. e Soybean Lipoxygenase-1. f Peptidylglycine α-hydroxylating monoxogyenase. g This work. Where necessary, reported changes in entropy of activation in e.u. were converted to unitless ln(AH/AD) by multiplication of both value and error by 0.503; changes in enthalpy of activation were used as reported for activation energy differences. | |||
| CoO + DHA | 3.7(3) | −3.3(5) | |
| CoO + Fluorene | 0.28(8) | 0.8(2) | |
| TAMLFeVO + EtPh | 26 | 3.3(8) | −4(2) |
| (TMP)+˙FeIV(O) + BnOH | 27 | 7.0(6) | −8(1) |
| LNHCFeIV(MeCN)(O)2+ + DHA | 28 | 4.5(5) | −5(1) |
| pipMeLCuIIIOH + DHA | 29 | 2.9(4) | −3(2) |
| LCuIIIOH + DHA | 29 | 0.4(2) | 3(3) |
| NO2LCuIIIOH + DHA | 29 | 3.6(3) | −5.0(7) |
| (LiPr3CuIII)2(μ-O)22+ab | 30 | 0.5(7) | 2.0(7) |
| 31 | 1.9(7) | −0.7(1) | |
| (LBn3CuIII)2(μ-O)22+a | 31 | 2.5(7) | −1.6(1) |
| (Tp′′CoII)2(μ-O2)a | 32 | 2.8(7) | −2.0(7) |
| RuIII(bpy)33+/H2O + PhOH | 33 | 1.68(4) | −1.40(7) |
| 4-oxo-TEMPO˙ + TEMPOHc | 34 | 0.3(6) | 1.1(6) |
| 9aH-Quinolozined | 35 | −0.01(2) | 1.64(3) |
| SLOe | 36 | 0.9(2) | 2.9(3) |
| PHMf | 37 | 0.4(3) | 1.8(5) |
KIEs for the oxidation of DHA are larger than 10 and have a temperature dependence significantly steeper than 1.4 kcal mol−1 (Fig. 1). This importantly suggests significant tunneling contributions in this reaction. It is consistent with either semiclassical theories with a tunneling correction, or with non-adiabatic rate theories with extensive tunneling. For instance, the reactivity of impaired enzymes often displays this behavior, and it is explained by invoking deuterium being more sensitive to tunneling distance.25 In any case, the KIE data for DHA clearly indicates there is significant proton tunnelling in the C–H activation reactions of CoO.
For fluorene, we instead observe a shallow slope and a significantly positive intercept of 0.8(2) in the plot of ln(KIE) vs. 1/RT. This indicates extensive tunnelling in this reaction, with proton and deuterium tunnelling being similarly efficient (Fig. 1). This observation supports that tunnelling is so extensive that semiclassical transition states are not sufficient (see ESI†). Much like in enzymatic systems, this behaviour can only be understood if the transferring proton is treated fundamentally as a quantum mechanical particle. This means that factors more typically thought of in the context of electron transfers, such as the effect of tunnelling distance and the reorganization energy, are central in understanding this reactivity.
The temperature insensitive KIEs for reactions between CoO and fluorene and the resulting positive value of ln(AH/AD) are uncommon in small molecule reactions; however, they are the norm in enzymatic systems (see Table 1 for a representative sample of other systems).10,26–37 Within the more specific context of metal complex mediated C–H activation, the paucity of data makes it difficult to draw general conclusions regarding the temperature dependence of KIEs and the importance of tunneling. Most reported data have negative intercept ln(AH/AD) values which suggest tunneling, but are technically still compatible with semiclassical theory.
In comparing CoO with other model complexes, our results are most reminiscent of a series of three copper hydroxide complexes’ reactivity with DHA.29 Two of these complexes display negative intercepts (ln(AH/AD) = −3(2) and −5.0(7)) but one displays a positive intercept (ln(AH/AD) = 3(3)). Similar results have been reported for bis-μ-oxo complexes of copper.30,31,38 It is not immediately apparent why reactivity between CoO and fluorene and some of these copper reactions are more “enzyme-like” with a positive intercept ln(AH/AD) whereas reactivity between CoO and DHA and most small molecule CPET reactions are not; however, these studies suggest that evidence for tunneling is perhaps best evaluated in a series of reactions rather than with just one substrate. As one general conclusion from our study, we note the need for additional variable temperature KIE data for molecular model complexes. Notably, many experimental studies use room temperature KIE values to determine whether tunneling is important. While room temperature KIE values >10 do mandate that tunneling is important, our study illustrates that values <10 can still indicate extensive tunneling.
The second major conclusion we draw is that the experimental evidence for extensive tunneling that we find in the reactivity of CoO with C–H bonds highlights the need to consider the quantum mechanical nature of the proton and non-adiabatic effects when explaining imbalanced reactivity trends in CPET; the use of semiclassical structure-reactivity relationships is not sufficient. Experimental demonstrations of imbalanced CPET must therefore be based on factors such as the tunneling distance, reorganization energy, and vibronic coupling. As an example, two recent studies found that lower PT energies correlated with shorter distances between the proton donor and the proton acceptor.39,40 The experimental data we present here bolsters recent questions about the compatibility of imbalanced thermodynamic driving forces with tunneling in CPET reactions.15–17
The need to properly address quantum mechanical tunneling more broadly within studies of transition metal-oxo mediated C–H activation also raises broader implications. For instance, many hypotheses regarding the acceleration of C–H activation by transition metal-oxo complexes have been supported primarily with DFT-calculated transition states which do not treat the proton as a quantum particle.3,20,41–43 Treating the proton quantum mechanically could blur the distinction between σ and π reaction pathways for metal-oxo mediated C–H activation. Extensive proton tunneling also complicates the consideration of spin states in metal oxo reactivity, as tunneling may circumvent spin-forbidden transitions.
In sum, we find that significant tunneling is present in the imbalanced CPET reactivity of CoOvia variable temperature KIE measurements. This experimentally supports recent calls to rationalize imbalanced CPET reactions with quantum mechanical proton tunnelling. Furthermore, this underscores the need for additional variable temperature KIE data and analysis on model complexes for CPET. More work is needed, both from theoretical and experimental chemists, to fully understand the role of tunneling in the reactivity of transition metal-oxo complexes.
This work was funded by the National Institutes of Health (R35 GM133470) and the University of Chicago. J. E. S. thanks the United States Department of Defense for a National Defense Science and Engineering Graduate Fellowship (00003765) and J. S. A. thanks the Sloan Foundation for a Research Fellowship (FG-2019-11497) and the Dreyfus Foundation for a Teacher-Scholar award (TC-21-064).
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Migliore, N. F. Polizzi, M. J. Therien and D. N. Beratan, Chem. Rev., 2014, 114, 3381–3465 CrossRef CAS PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- D. Bím, M. Maldonado-Domínguez, L. Rulíšek and M. Srnec, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E10287–E10294 CrossRef.
- J. W. Darcy, S. S. Kolmar and J. M. Mayer, J. Am. Chem. Soc., 2019, 141, 10777–10787 CrossRef CAS PubMed.
- S. K. Barman, J. R. Jones, C. Sun, E. A. Hill, J. W. Ziller and A. S. Borovik, J. Am. Chem. Soc., 2019, 141, 11142–11150 CrossRef CAS.
- M. K. Goetz and J. S. Anderson, J. Am. Chem. Soc., 2019, 141, 4051–4062 CrossRef CAS.
- D. Usharani, D. C. Lacy, A. S. Borovik and S. Shaik, J. Am. Chem. Soc., 2013, 135, 17090–17104 CrossRef CAS.
- C. E. Elwell, M. Mandal, C. J. Bouchey, L. Que, C. J. Cramer and W. B. Tolman, Inorg. Chem., 2019, 58, 15872–15879 CrossRef CAS PubMed.
- M. Mandal, C. E. Elwell, C. J. Bouchey, T. J. Zerk, W. B. Tolman and C. J. Cramer, J. Am. Chem. Soc., 2019, 141, 17236–17244 CrossRef CAS PubMed.
- S. K. Barman, M.-Y. Yang, T. H. Parsell, M. T. Green and A. S. Borovik, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(36), e2108648118 CrossRef CAS PubMed.
- J. E. Schneider, M. K. Goetz and J. S. Anderson, Chem. Sci., 2021, 12, 4173–4183 RSC.
- J. M. Hodgkiss, J. Rosenthal and D. G. Nocera, in Hydrogen-Transfer Reactions, ed. J. T. Hynes, J. P. Klinman, H.-H. Limbach and R. L. Schowen, John Wiley & Sons, Ltd, 2006, pp. 503–562 Search PubMed.
- J. K. Bower, A. D. Cypcar, B. Henriquez, S. C. E. Stieber and S. Zhang, J. Am. Chem. Soc., 2020, 142, 8514–8521 CrossRef CAS PubMed.
- H. Kotani, H. Shimomura, K. Ikeda, T. Ishizuka, Y. Shiota, K. Yoshizawa and T. Kojima, J. Am. Chem. Soc., 2020, 142, 16982–16989 CrossRef CAS PubMed.
- C. Costentin and J.-M. Savéant, Chem. Sci., 2020, 11, 1006–1010 RSC.
- R. Tyburski, T. Liu, S. D. Glover and L. Hammarström, J. Am. Chem. Soc., 2021, 143, 560–576 CrossRef CAS PubMed.
- T. Liu, R. Tyburski, S. Wang, R. Fernández-Terán, S. Ott and L. Hammarström, J. Am. Chem. Soc., 2019, 141, 17245–17259 CrossRef CAS PubMed.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Mill Valley, California, 2006 Search PubMed.
- R. P. Bell, The tunnel effect in chemistry, Chapman and Hall, London, New York, 1980 Search PubMed.
- D. Mandal, D. Mallick and S. Shaik, Acc. Chem. Res., 2018, 51, 107–117 CrossRef CAS PubMed.
- R. I. Cukier and D. G. Nocera, Annu. Rev. Phys. Chem., 1998, 49, 337–369 CrossRef CAS PubMed.
- S. Hammes-Schiffer, Energy Environ. Sci., 2012, 5, 7696–7703 RSC.
- S. Hammes-Schiffer and A. A. Stuchebrukhov, Chem. Rev., 2010, 110, 6939–6960 CrossRef CAS PubMed.
- J. P. Klinman, J. Phys. Org. Chem., 2010, 23, 606–612 CrossRef CAS.
- J. P. Klinman and A. R. Offenbacher, Acc. Chem. Res., 2018, 51, 1966–1974 CrossRef CAS PubMed.
- S. Kundu, J. V. K. Thompson, L. Q. Shen, M. R. Mills, E. L. Bominaar, A. D. Ryabov and T. J. Collins, Chem. – Eur. J., 2015, 21, 1803–1810 CrossRef CAS PubMed.
- Z. Pan, J. H. Horner and M. Newcomb, J. Am. Chem. Soc., 2008, 130, 7776–7777 CrossRef CAS PubMed.
- C. Kupper, B. Mondal, J. Serrano-Plana, I. Klawitter, F. Neese, M. Costas, S. Ye and F. Meyer, J. Am. Chem. Soc., 2017, 139, 8939–8949 CrossRef CAS PubMed.
- D. Dhar, G. M. Yee, A. D. Spaeth, D. W. Boyce, H. Zhang, B. Dereli, C. J. Cramer and W. B. Tolman, J. Am. Chem. Soc., 2016, 138, 356–368 CrossRef CAS PubMed.
- S. Mahapatra, J. A. Halfen, E. C. Wilkinson, L. Que and W. B. Tolman, J. Am. Chem. Soc., 1994, 116, 9785–9786 CrossRef CAS.
- S. Mahapatra, J. A. Halfen and W. B. Tolman, J. Am. Chem. Soc., 1996, 118, 11575–11586 CrossRef CAS.
- O. M. Reinaud and K. H. Theopold, J. Am. Chem. Soc., 1994, 116, 6979–6980 CrossRef.
- J. Bonin, C. Costentin, C. Louault, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2011, 133, 6668–6674 CrossRef CAS PubMed.
- A. Wu, E. A. Mader, A. Datta, D. A. Hrovat, W. T. Borden and J. M. Mayer, J. Am. Chem. Soc., 2009, 131, 11985–11997 CrossRef CAS PubMed.
- H. Kwart, M. W. Brechbiel, R. M. Acheson and D. C. Ward, J. Am. Chem. Soc., 1982, 104, 4671–4672 CrossRef CAS.
- M. P. Meyer, D. R. Tomchick and J. P. Klinman, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1146–1151 CrossRef CAS PubMed.
- W. A. Francisco, M. J. Knapp, N. J. Blackburn and J. P. Klinman, J. Am. Chem. Soc., 2002, 124, 8194–8195 CrossRef CAS PubMed.
- S. Mahapatra, J. A. Halfen, E. C. Wilkinson, G. Pan, C. J. Cramer, L. Que and W. B. Tolman, J. Am. Chem. Soc., 1995, 117, 8865–8866 CrossRef CAS.
- E. R. Sayfutyarova, Y.-C. Lam and S. Hammes-Schiffer, J. Am. Chem. Soc., 2019, 141, 15183–15189 CrossRef CAS PubMed.
- R. Tyburski and L. Hammarström, Chem. Sci., 2021, 13, 290–301 RSC.
- D. Mandal and S. Shaik, J. Am. Chem. Soc., 2016, 138, 2094–2097 CrossRef CAS PubMed.
- W. Lai, C. Li, H. Chen and S. Shaik, Angew. Chem., Int. Ed., 2012, 51, 5556–5578 CrossRef CAS PubMed.
- K.-B. Cho, E. J. Kim, M. S. Seo, S. Shaik and W. Nam, Chem. – Eur. J., 2012, 18, 10444–10453 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols, detailed summary of KIE data, and graphs of kinetic traces and GC-MS data. See DOI: https://doi.org/10.1039/d3cc02130k |
| This journal is © The Royal Society of Chemistry 2023 |