
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A thirty-year old mystery solved: identification of a new heptatungstate from non-aqueous solutions†
Dominic
Shiels
 a,
Magda
Pascual-Borràs
a,
Paul G.
Waddell
a,
Corinne
Wills
a,
Josep-Maria
Poblet
b and
R. John
Errington
*a
a,
Magda
Pascual-Borràs
a,
Paul G.
Waddell
a,
Corinne
Wills
a,
Josep-Maria
Poblet
b and
R. John
Errington
*a
aChemistry, School of Natural & Environmental Sciences, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: John.Errington@newcastle.ac.uk
bDepartament de Quimica Fisica i Inorgànica, Universitat Rovira i Virgili, Marcel·lí Domingo 1, Tarragona 43007, Spain
First published on 31st May 2023
Abstract
A new isopolyoxotungstate has been characterised, thirty years since the first spectroscopic evidence of its existence. The heptatungstate [W7O24H]5−, containing a {W5} lacunary Lindqvist unit fused to a ditungstate fragment, has significant stability and is only the third isopolytungstate structure to be obtained from non-aqueous systems.
The formation of isopolyoxotungstates [WxOyHz]n− by acidification of aqueous [WO4]2− solutions is well established.1 Anions with different n/x values may be isolated from such solutions and characterised by single crystal X-ray diffraction, but these do not necessarily provide a true indication of the structures present in solution and, in this regard, the detailed 183W and 17O NMR studies of Maksimovskaya and Howarth were pivotal in developing an understanding of the pH-dependent speciation in aqueous isopolytungstate solutions.2,3 Recently, Falaise and co-workers have demonstrated that supramolecular interactions in the presence of γ-cyclodextrin can have a profound effect on the tungstate species present in such solutions.4 Details of isopolytungstate formation in non-aqueous media have been far less investigated and, importantly, the influence of water on the formation and solution stability of isopolytungstate structures in organic media is poorly understood. Although organic-soluble tetraalkylammonium salts of isopolytungstates have been characterised by liquid state 183W and 17O NMR spectroscopy, these are generally prepared from aqueous solutions by cation exchange,1 so these studies provide limited insight into the effects on the assembly process of moving from aqueous to organic solvents. An exception is the seminal work of Jahr and co-workers, who prepared polyoxometalates (POMs) by basic hydrolysis of metal alkoxides in organic solvents, including the synthesis of [W6O19]2− from the oxoalkoxides WO(OR)4 (R = Me, Et).5
In 1993, we reported 183W NMR studies of non-aqueous attempts to prepare the unknown molecular ditungstate [W2O7]2− as its nBu4N+ (TBA) salt, including the hydrolytic approach shown in eqn (1).6 These revealed an intriguing and apparently new isopolytungstate, but its high solubility has frustrated all subsequent attempts at crystallographic characterisation. Our non-aqueous studies of heterometal-containing {MW5} Lindqvist-type POMs have involved syntheses from putative lacunary precursors [W5O18Hz](6−z)−, targeted either by the controlled hydrolysis of WO(OMe)4 in the presence of [WO4]2− as in eqn (2) and (3) or, more recently, by base-degradation of [W6O19]2− as in eqn (4) and (5). Both approaches facilitate 17O enrichment of the POM framework, either by addition of 17O enriched water or by degradation of 17O-enriched [W6O19]2−. Direct treatment of these dynamic reaction mixtures with heterometal precursors M(OR)4 (z = 3) or MX2 (z = 1) provides versatile access to a series of reactive Lindqvist polyoxometalates [(RO)MW5O18]3− or [(MW5O18H)2]6− respectively.7–11
| [WO4]2− + WO(OMe)4 + 2 H2O → [W2O7]2− + 4 MeOH | (1) |
| 3 [WO4]2− + 7 WO(OMe)4 + 17 H2O → 2 “[W5O18H3]3−” + 28 MeOH | (2) |
| 5 [WO4]2− + 5 WO(OMe)4 + 11 H2O → 2 “[W5O18H]5−” + 20 MeOH | (3) |
| 5 [W6O19]2− + 8 OH− + 5 H2O → 6 “[W5O18H3]3−” | (4) |
| 5 [W6O19]2− + 20 OH− → 6 “[W5O18H]5−” + 7 H2O | (5) |
In attempts to identify the species generated in non-aqueous reactions targeting “virtual” lacunary [W5O18Hz](6−z)− precursors, we have examined the solutions by 183W and 17O NMR spectroscopy. Degradation of [W6O19]2− with the stoichiometry in eqn (5), i.e. where z = 1 and n/x = 1.0, followed by removal of the volatiles gave a product with the 17O NMR spectrum shown in Fig. S1 (ESI†), where the two sets of broad peaks 721–642 ppm and 395–328 ppm are characteristic of terminal W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and bridging WOW respectively. The peaks at 437 and −1.6 ppm are assigned to [WO4]2− and H2O respectively. The 183W NMR spectrum of this solution shown in Fig. 1 contains a peak at −5 ppm due to [WO4]2− and five peaks at 53, 50, 38, −59 and −77, ppm in the ratio 1
O and bridging WOW respectively. The peaks at 437 and −1.6 ppm are assigned to [WO4]2− and H2O respectively. The 183W NMR spectrum of this solution shown in Fig. 1 contains a peak at −5 ppm due to [WO4]2− and five peaks at 53, 50, 38, −59 and −77, ppm in the ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1:1:2 respectively, which is indicative of an isopolytungstate containing 7n tungsten atoms in five unique environments. These spectra are remarkably similar to those obtained during our attempts to prepare [W2O7]2− by reaction (1),6 although peaks in previous 183W NMR spectra were ∼ 9 ppm upfield of those in the current studies. This further indicated that the 5-line 183W NMR spectrum with 1:2:1:1:2 peak intensities is associated with a fundamental polytungstate structure in non-aqueous solutions of [WxOyHz]n− with n/x = 1.0.
:2:1:1:2 respectively, which is indicative of an isopolytungstate containing 7n tungsten atoms in five unique environments. These spectra are remarkably similar to those obtained during our attempts to prepare [W2O7]2− by reaction (1),6 although peaks in previous 183W NMR spectra were ∼ 9 ppm upfield of those in the current studies. This further indicated that the 5-line 183W NMR spectrum with 1:2:1:1:2 peak intensities is associated with a fundamental polytungstate structure in non-aqueous solutions of [WxOyHz]n− with n/x = 1.0.
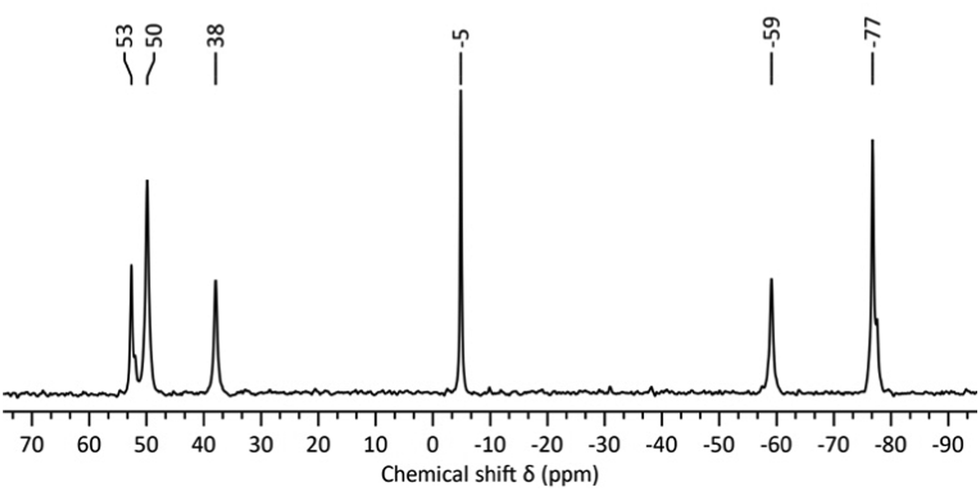 | ||
| Fig. 1 183W NMR spectrum of the product from treatment of (TBA)2[W6O19] with 4 equivalents of (TBA)OH in MeCN. | ||
Details of our extensive investigations into non-aqueous tungstate speciation will be reported elsewhere, but 17O and 183W NMR spectra from the degradation of (TBA)2[W6O19] with varying amounts of TBA(OH) are shown in Fig. S2–S11 (ESI†). Note that the spectrum of (TBA)2[W6O19] (Fig. S2, ESI†) was recorded with a significantly longer delay time between pulses (600 seconds) to enable observation of the central oxygen at −79 ppm. This oxygen has an extremely long T1 relaxation time of ∼ 55(±1) s due to the absence of an electric field gradient and associated relaxation mechanism for the quadrupolar 17O in the centre of the highly symmetrical [W6O19]2− anion. After addition of 0.4 mole-equivalent of TBA(OH) to (TBA)2[W6O19] a complex 17O NMR spectrum was observed with several broad peaks in both terminal WO and bridging W–O–W regions. The more intense, narrow peaks at 416 ppm and 776 ppm are characteristic of [W6O19]2−, indicating incomplete degradation of the hexatungstate. With increasing amounts of base, the spectra simplified somewhat as the residual [W6O19]2− was consumed, while the peak assigned to [WO4]2− at 437–439 ppm steadily increased in intensity with a concomitant decrease in the intensity of the broad WO and W–O–W features. The complexity of 17O NMR spectra prevented definitive peak assignments for the species responsible for the 183W NMR spectrum shown in Fig. 1.
Vapour diffusion of diethyl ether into the NMR solution for the reaction between (TBA)2[W6O19] and 1 mole-equivalent of TBA(OH) to give n/x = 0.5 produced colourless crystals over several weeks. Single-crystal X-ray crystallographic analysis showed these to be a 1:1 co-crystalline mixture of [W6O19]2− and the new isopolytungstate [W7O24H]5−1 shown in Fig. 3. The average n/x value in the crystal is 0.53 and the full structure is shown in Fig. S13 (ESI†). The protonated formula for 1 is supported by the number of TBA cations in the crystal structure of (TBA)71·[W6O19]·3MeCN, the hydrogen-bonded O17–O20 distance and the observation of a peak at 8.51 ppm in the 1H NMR spectrum (Fig. S12, ESI†). The structure of 1 is markedly different from that of the heptatungstate [W7O24]6− obtained from aqueous solutions, and can be regarded as a fusion of lacunary {W5O18} and ditungstic {W2O5OH} fragments, a feature similar to that observed in (W2}-capped lacunary {BW11O39} units isolated from aqueous borotungstate solutions.12 Using Pope's classification, 1 can be described as a type III POM containing addenda atoms with both one or two terminal MO bonds.13 The longer terminal WO bond lengths in the W5 unit of 1 (W1–W4, average ca. 1.73 Å) compared with those for the [W6O19]2− anion in the co-crystal (average ca. 1.69 Å) can be ascribed to the higher charge associated with 1. The bridging W–O bond lengths in 1 show significant distortions compared with those in [W6O19]2− (average bridging W–O of ca. 1.92 Å). The asymmetric links between the {W5} and {W2} units in 1 have WOW angles of 146.8° and 146.1°, with shorter W–O bonds to the {W5} unit (W2–O15, W3–O16 and W5–O18, average ca. 1.88 Å), while the lengthening of W2–O2, W3–O3, W4–O4 and W5–O5 bonds within the {W5} unit (average ca.1.99 Å) is consistent with a trans effect from more strongly π-bonding oxygens. Bridging W–O bonds in the {W5} equatorial plane and those to W1 are similar to those observed in [W6O19]2−, with an average of ca. 1.92 Å. Bond valence sum analysis (Table S3, ESI†) indicates localisation of the proton on O20 rather than on O17 (VO20 = 1.18 vs. VO17 = 1.58). Together with the short W4–O17 bond length of 1.750(7) Å, indicating significant WO π-bonding, this implies that this linkage is best described as WO⋯HOW2 rather than WOH⋯OW2 involving terminal W–OH. The capping {W2} unit contains two cis-WO2 fragments with average terminal WO bond lengths of ca. 1.74 Å, which is similar to the others in the structure. For the three oxygens bridging W6 and W7, the shortest W–O bonds (1.940(7) and 1.949(7) Å) are to O23, which is μ2-O, while those to O20 (μ2-OH) and O15 (μ3-O) average ca. 2.20 Å. The effective Cs symmetry of 1 gives a ratio of 1:2:1:1 for the five tungsten atoms in the {W5} unit, with equivalent W atoms in the {W2} capping unit. This is consistent with the 5-line pattern of the 183W NMR spectrum in Fig. 1, and also with the 2JWW coupling previously observed for the two most intense peaks, as 2JWW values of ∼ 20 Hz are associated with larger WOW angles.14 The numbers of WO and WOW peaks observed in 17O NMR spectra of products from reactions (1) and (5) are also consistent with the structure in Fig. 3.
Density functional theory was used to optimise the structure of [W7O24H]5− and calculate 183W NMR parameters, including 2JWW between W3/W5 and W6/W7 (ESI†). The computed 183W chemical shifts obtained from different computational procedures are shown in Table 1 and deviations from the experimental values obtained in this study are given as mean absolute errors (MAEs). The best methodology for reproducing the experimental 183W NMR spectrum of 1 is OPBE/TZP//PBE/TZ2P, with a MAE of 34 ppm compared to the values shown in Fig. 1. Calculated values for 2JWW coupling between W3/W5 and W6/W7 are in excellent agreement with our previous experimental values,6 regardless of the methodology used. 17O NMR chemical shifts for 1 were also computed (Table S4, ESI†) in order to rationalise the series of peaks observed in the terminal WO and bridging W–O–W regions of the 17O NMR spectrum and results were consistent with the overlapping pattern of peaks shown in Fig. S1 (ESI†).
| Procedure (NMR//OPT) | Chemical shift/ppm | MAE | 2 J WW/Hz | ||||
|---|---|---|---|---|---|---|---|
| W1 | W4 | W5/3 | W2 | W6/7 | |||
| a Value taken from ref. 6. | |||||||
| PBE/TZP//PBE86/TZ2P | −44 | 138 | 152 | 118 | −38 | 64 | 22.6 |
| OPBE/TZP//PBE/TZ2P | −98 | 93 | 105 | 70 | −80 | 34 | 22.8 |
| PBE/TZP//OPBE/TZ2P | −66 | 94 | 126 | 87 | −60 | 38 | 22.8 |
| BP86/TZP//BP86/QZ4P | −51 | 135 | 146 | 112 | −46 | 58 | 23.0 |
| Observed | −59 | 53 | 50 | 38 | −77 | — | 22.6a |
Despite the minor variations in 183W chemical shifts for different samples, this led us to believe that anion 1 was indeed responsible for the 5-line 183W and complex 17O NMR spectra observed for reactions (1) and (5). To shed light on the chemical shift variations, eliminate ambiguity from spectral assignments and confirm the presence of [WO4]2−, 183W NMR studies of the degradation mixture from (5) after removal of the volatiles were repeated with a capillary insert containing 2 M Na2WO4·2H2O in D2O (Fig. 2a), which provided a consistent reference peak at 0.5 ppm. The upfield peak for 1 at ∼ −75 ppm showed the greatest chemical shift variation of ca. 4 ppm but most striking was the ca. 20 ppm variation in the [WO4]2− chemical shift (Fig. 2a). The effects of concentration and the presence of water on the chemical shift of [WO4]2− were therefore investigated. 183W NMR spectra of 2 M and 1 M solutions of (TBA)2[WO4] in MeCN (Fig. 2b and c) showed an upfield shift of 2 ppm for the 1 M solution but, as the reaction mixtures used for the 183W NMR spectra shown in Fig. 1b and 2a contained similar relative amounts of [WO4]2−, concentration did not appear to be the cause of the large chemical shift difference. More significantly, the addition of one equivalent of water to the 1 M solution of (TBA)2[WO4] in MeCN (Fig. 2d) produced an upfield shift of >9 ppm. Given that water is an expected product from base-degradation of [W6O19]2−, it is likely that this is the cause of the variation in the [WO4]2− peak position, which is not surprising in view of the high basicity of the [WO4]2− anion as exemplified by the hydrogen-bonded water in the crystal structure of (BTMA)2[WO4]·H2O (BTMA = PhCH2Me3N+).15 The presence of H2O or MeOH might similarly be expected to affect chemical shifts of W atoms bonded to basic oxygens in 1.
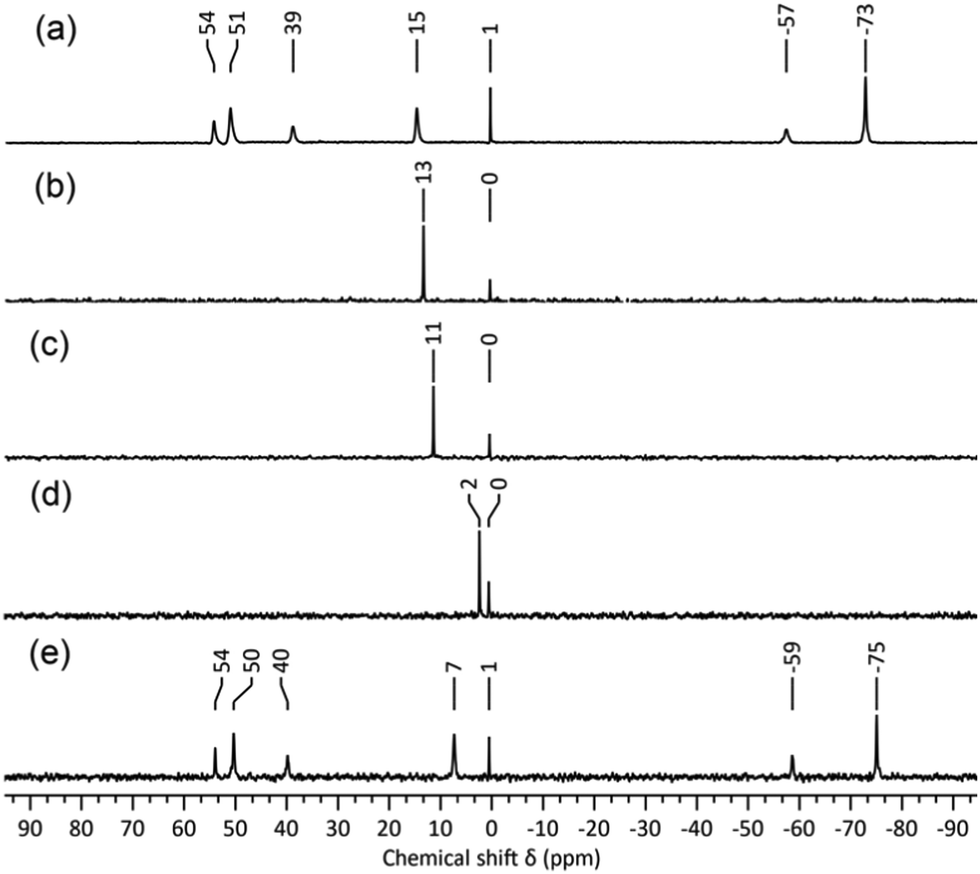 | ||
| Fig. 2 183W NMR spectra of (a) (TBA)2[W6O19] + 4 eq. of TBA(OH), (b) 2 M (TBA)2[WO4], (c) 1 M (TBA)2[WO4], (d) 1 M (TBA)2[WO4] + 1 eq. H2O, and (e) a combination of solutions responsible for the spectra in (a) and (d). All spectra recorded in MeCN with an internal co-axial capillary containing 2 M Na2WO4·2H2O in D2O. | ||
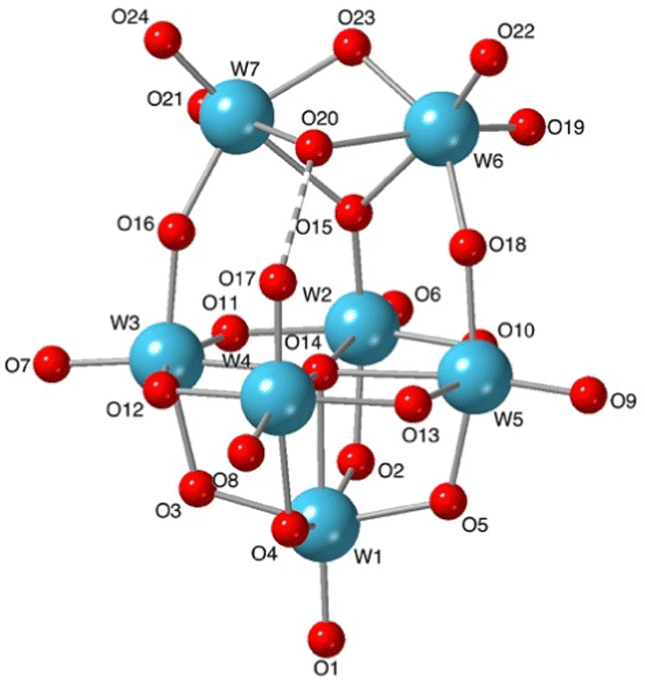 | ||
| Fig. 3 Structure of [W7O24H]5−1 in the co-crystal (TBA)71·[W6O19]·3MeCN. | ||
It was also evident that the linewidth of the [WO4]2− peak is significantly broader in reaction mixtures produced by base-degradation of (TBA)2[W6O19] than in spectra of (TBA)2[WO4] alone. The FWHM increases from ca. 2.5 Hz in Fig. 2b–d to ca. 10.5 Hz in spectra of solutions that also contain 1. This was confirmed by combining the reaction mixture responsible for the 183W spectrum in Fig. 2a with the 1 M (TBA)2[WO4] solution containing one equivalent of water. In the 183W NMR spectrum of the resulting mixture (Fig. 2e), the chemical shift of [WO4]2− is approximately the mean of the corresponding shifts in the original solutions, which may be explained by the lowered H2O: [WO4]2− ratio in the mixture. The broadened [WO4]2− peak in Fig. 1 and 2a compared with that for the 1 M solution of (TBA)2[WO4] with 1 eq. of water also suggests exchange between [WO4]2− and 1. Peaks in the 183W NMR spectra of 1 from our previous studies were notably narrower than those from these current studies, and a 2JWW coupling of 22.6 Hz was resolved from satellites associated with the larger peaks now assigned to W(3/5) and W(6/7). The absence of resolved coupling in Fig. 1 and 2a may be explained by either (i) exchange involving [WO4]2− formed in base degradation reactions or (ii) line broadening due to chemical shift anisotropy, which is expected to be more problematic with the low-gamma broad-band probe and 11.7 T magnet than with the dedicated 183W probe and 7.05 T magnet used in previous studies.
While these combined NMR, structural and computational data suggested strongly that 1 is the main species produced in non-aqueous tungstate solutions with n/x = 1.0, insufficient (TBA)71.·[W6O19]·3MeCN co-crystals were obtained to record confirmatory 183W and 17O NMR spectra and we sought to obtain X-ray structural data on crystals of a salt of 1 obtained from a solution with the characteristic 5-line 183W NMR spectrum. The high solubility in organic solvents of (TBA)51 obtained from reactions (1) and (5) precluded its crystallisation and separation from any (TBA)2[WO4] formed in degradation reactions. In order to isolate crystals containing 1 in the absence [W6O19]2−, we therefore treated (TBA)2[W6O19] with four mole-equivalents of (BTMA)OH in a mixture of MeOH and MeCN. The white precipitate obtained after stirring at room temperature overnight was recrystallised from hot DMSO/DMF to give a mixture of amorphous solid and colourless crystals, which were shown to be (BTMA)5[W7O24H]·2DMSO·1.71H2O by single-crystal X-ray diffraction (Fig. S14, ESI†). FTIR spectra of the crystalline material and the amorphous solid were identical (Fig. S17 and S18, ESI†), but we were unable to record a 183W NMR spectrum of (BTMA)51 due to its low solubility in organic solvents. To prove that the 5-line 183W NMR spectra obtained from reactions (1) and (5) were both due to 1 we first carried out reaction (5) in MeCN to obtain a spectrum analogous to that shown in Fig. 1, then removed the solvent and re-recorded the 183W NMR spectrum in DMSO to confirm the retention of a 5-line spectrum (Fig. S10 and S11, ESI†). Subsequent addition of (BTMA)Br and recrystallisation of the resulting precipitate from hot DMSO/DMF gave crystals that were shown by X-ray crystallography to be (BTMA)51·2DMSO by comparison of unit cell parameters with the previous sample (Table S2, ESI†). Elemental microanalysis was consistent with a formula having five BTMA cations and hence a protonated anion.
To our knowledge, 1 joins [W6O19]2− and [W10O32]4− as the only isopolytungstates to be isolated and characterised from non-aqueous solutions. In the FTIR spectrum of (BTMA)51 (Fig. S17, ESI†), the band for ν(WO) at 922 cm−1 is lower than that for (TBA)2[W6O19] and (TBA)4[W10O32] at 967 and 953 cm−1 respectively due to the greater anionic charge of 1. By analogy with the FTIR spectra of (TBA)2[Mo6O19] and (TBA)2[Mo2O7], either of the bands at 863 or 821 cm−1 may be associated with ν(WO) for cis-WO2 in 1.
We have demonstrated conclusively that the new isopolytungstate [W7O24H]5−1 is formed by both hydrolytic aggregation reaction (1) and by degradation reaction (5). Crucially, this means that 1 is present in non-aqueous [WxOyHz]n− mixtures with n/x = 1.0, i.e. those used in our attempts to prepare (TBA)2[W2O7] or lacunary Lindqvist-type (TBA)5[W5O18H] species, and for the synthesis of a wide range of heterometal-substituted Lindqvist {MW5O18} anions, suggesting that the {W5} fragment is retained upon treatment with a wide variety of heterometal sources. This represents a major advance in the understanding of previously ill defined “virtual” precursor solutions and will further guide our development of rational protocols for targeted POM synthesis.
We are grateful to EPSRC for support through the RENU CDT (EP/S023836/1) and the INPOMS UK-Japan Core-to-Core Network Grant (EP/S031170/1).
Conflicts of interest
There are no conflicts to declare.References
- (a) M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer-Verlag, Berlin, 1983 CrossRef; (b) J. J. Cruywagen, Adv. Inorg. Chem., 2000, 49, 127–182 Search PubMed; (c) W. G. Klemperer, Early transition metal polyoxoanions, in Inorganic Syntheses, ed. A. P. Ginsberg, John Wiley & Sons, 1990, 27, pp. 71–135 Search PubMed; (d) G. Hervé, A. Tézé and R. Contant, Polyoxometalate Molecular Science, Springer Netherlands, Dordrecht, 2003, 33–54 Search PubMed.
- R. I. Maksimovskaya and K. G. Burtseva, Polyhedron, 1985, 4, 1559–15623 CrossRef CAS.
- J. J. Hastings and O. W. Howarth, J. Chem. Soc., Dalton Trans., 1992, 209–215 RSC.
- C. Falaise, M. A. Moussawi, S. Floquet, P. A. Abramov, M. N. Sokolov, M. Haouas and E. Cadot, J. Am. Chem. Soc., 2018, 140, 11198–11201 CrossRef CAS PubMed.
- (a) K. F. Jahr and J. Fuchs, Angew. Chem., Int. Ed. Engl., 1996, 9, 689–750 Search PubMed; (b) K. F. Jahr and J. Fuchs, Chem. Ber., 1963, 96, 2457–2459 CrossRef CAS; (c) K. F. Jahr, J. Fuchs and R. Oberhauser, Chem. Ber., 1968, 101, 477–481 CrossRef CAS.
- R. J. Errington, M. D. Kerlogue and D. G. Richards, J. Chem. Soc., Chem. Commun., 1993, 649–651 RSC.
- W. Clegg, M. R. J. Elsegood, R. J. Errington and J. Havelock, J. Chem. Soc., Dalton Trans., 1996, 681–690 RSC.
- R. J. Errington, S. S. Petkar, P. S. Middleton, W. McFarlane, W. Clegg, R. A. Coxall and R. W. Harrington, J. Chem. Soc., Dalton Trans., 2006, 5211–5222 Search PubMed.
- R. J. Errington, S. S. Petkar, P. S. Middleton, W. McFarlane, W. Clegg, R. A. Coxall and R. W. Harrington, J. Am. Chem. Soc., 2007, 129, 12181–12196 CrossRef CAS PubMed.
- R. J. Errington, G. Harle, W. Clegg and R. W. Harrington, Eur. J. Inorg. Chem., 2009, 5240–5246 CrossRef CAS.
- B. Kandasamy, C. Wills, W. McFarlane, W. Clegg, R. W. Harrington, A. Rodríguez-Fortea, J. M. Poblet, P. G. Bruce and R. J. Errington, Chem. – Eur. J., 2012, 18, 59–62 CrossRef CAS PubMed.
- (a) A. Tézé, M. Michelon and G. Hervé, Inorg. Chem., 1997, 36, 505–509 CrossRef; (b) N. Leclerc-Laronze, J. Marrot, G. Hervé, R. Touvenot and E. Cadot, Chem. – Eur. J., 2007, 13, 7234–7245 CrossRef CAS PubMed.
- M. T. Pope, Inorg. Chem., 1972, 11, 1973–1974 CrossRef CAS.
- (a) J. Lefebvre, F. Chauveau, P. Doppelt and C. Brevard, J. Am. Chem. Soc., 1981, 103, 4589–4591 CrossRef CAS; (b) W. H. Noth, P. J. Domaille and D. C. Roe, Inorg. Chem., 1983, 22, 198–201 CrossRef; (c) C. Brevard, R. Schimpf, G. Tourné and C. M. Tourné, J. Am. Chem. Soc., 1983, 105, 7059–7063 CrossRef CAS.
- W. Clegg, R. J. Errington, K. A. Fraser and D. G. Richards, J. Chem. Soc., Chem. Commun., 1993, 1105–1107 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, NMR, computational and crystallographic details. CCDC 2259327 and 2259328. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc02061d |
| This journal is © The Royal Society of Chemistry 2023 |