
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modular synthesis of bicyclic twisted amides and anilines†
Alexandra
Hindle
a,
Krzysztof
Baj
 b,
Jonathan A.
Iggo
b,
Daniel J.
Cox
c,
Christopher M.
Pask
a,
Adam
Nelson
*ad and
Stephen P.
Marsden
*a
b,
Jonathan A.
Iggo
b,
Daniel J.
Cox
c,
Christopher M.
Pask
a,
Adam
Nelson
*ad and
Stephen P.
Marsden
*a
aSchool of Chemistry, University of Leeds, Leeds, LS2 9JT, UK. E-mail: s.p.marsden@leeds.ac.uk
bDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool L69 7ZD, UK
cRedbrick Molecular, The Innovation Centre, 217 Portobello, Sheffield, S1 4DP, UK
dAstbury Centre for Structural Molecular Biology, University of Leeds, Leeds, LS2 9JT, UK. E-mail: a.s.nelson@leeds.ac.uk
First published on 17th April 2023
Abstract
Bridged amides and anilines display interesting properties owing to perturbation of conjugation of the nitrogen lone-pair with the adjacent π-system. A convergent approach to diazabicyclic scaffolds which contain either twisted amides or anilines is described, based on the photocatalysed hydroamination of cyclic enecarbamates and subsequent cyclisation. The modular nature of the synthesis allows for variation of the degree of ‘twist’ and hence the properties of the amides and anilines.
The impact of resonance upon both the structure and chemical properties of amides has been recognised since Pauling's seminal treatise on the nature of the chemical bond.1 Maximisation of this resonance requires planarity of the amide, and while small departures from this may be tolerated without significant energetic penalty (e.g. in many peptidic structures),2 larger deviations lead to a significant reduction in nN to π*C=O conjugation, and hence unusual structural and chemical properties. Twisted amides have therefore received significant attention as chemists seek to understand and exploit their unique characteristics and reactivity profiles.3 A prominent example is Kirby's most-twisted amide 1 (Fig. 1, panel a), in which the nitrogen lone pair is perpendicular to the carbonyl π-bond: the resulting lack of resonance manifests in ‘aminoketone’-like behaviour structurally (lengthened C–N and shortened C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds), spectroscopically (δC = 200 ppm for CO), and in reactivity (rapid hydrolysis at room temperature).4 More recently, the landmark synthesis of 2-quinuclidonium salts such as 2 by Stoltz5 has enabled experimental quantification of the high basicity of the nitrogen lone pair,5b which contrasts with the greater basicity on oxygen exhibited by simple amides. While compounds such as 1 and 2 represent the extremes of behaviour of twisted amides, the ability to tune molecular properties such as the relative basicities6 and hydrogen-bonding capabilities of the oxygen and nitrogen atoms across families of homologous amides is a source of fascination and utility to chemists.
O bonds), spectroscopically (δC = 200 ppm for CO), and in reactivity (rapid hydrolysis at room temperature).4 More recently, the landmark synthesis of 2-quinuclidonium salts such as 2 by Stoltz5 has enabled experimental quantification of the high basicity of the nitrogen lone pair,5b which contrasts with the greater basicity on oxygen exhibited by simple amides. While compounds such as 1 and 2 represent the extremes of behaviour of twisted amides, the ability to tune molecular properties such as the relative basicities6 and hydrogen-bonding capabilities of the oxygen and nitrogen atoms across families of homologous amides is a source of fascination and utility to chemists.
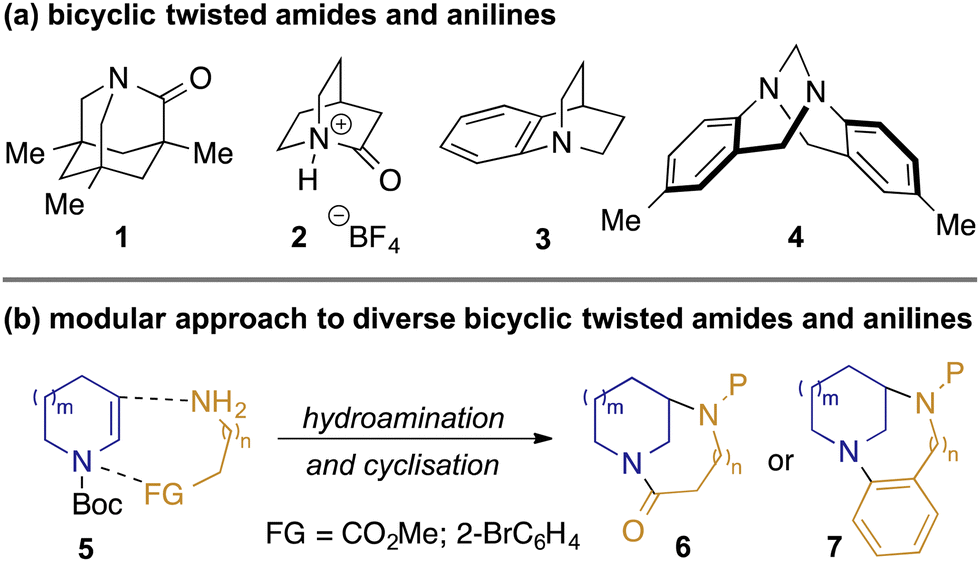 | ||
| Fig. 1 Bicyclic twisted amides and anilines, and a proposed modular approach to congeners. | ||
While less widely studied than amides, similar effects are observed with twisted anilines such as benzoquinuclidine 37 and Tröger's base 4,8 in which conjugation between the nitrogen lone pair and aromatic group are disrupted. The orthogonality of the nitrogen lone-pair with the arene in 3 manifests itself, for example, in the anomalous basicity of the amine (pKa = 7.8, c.f. 5.2 for dimethylaminobenzene),9 the observation of meta-substitution in SEAr reactions under acidic conditions,10 and in anomalous photophysical behaviour of donor-acceptor compounds containing this motif.11
Previously we reported the preparation of diazabicyclic twisted amides based on the bicyclo[3.3.1]nonane and bicyclo[4.3.1]decane scaffolds.12 Our synthesis employed reductive amination of 3-ketoazacycles with α- and β-aminoesters to convergently prepare precursors for Bu2SnO-mediated lactamisation. Although successful, this approach limited exploration e.g. of alternative ring-sizes and substitution patterns. We describe herein a complementary and more general approach, based upon coupling of key building blocks through photoredox-mediated hydroamination13,14 of readily-available15 enecarbamates 5 (Scheme 1, panel b). Cyclisation either through lactamisation or Buchwald–Hartwig amination gives a broad range of bicyclic twisted amides 6 and anilines 7 wherein the impact of subtle changes in structure upon properties can be probed.
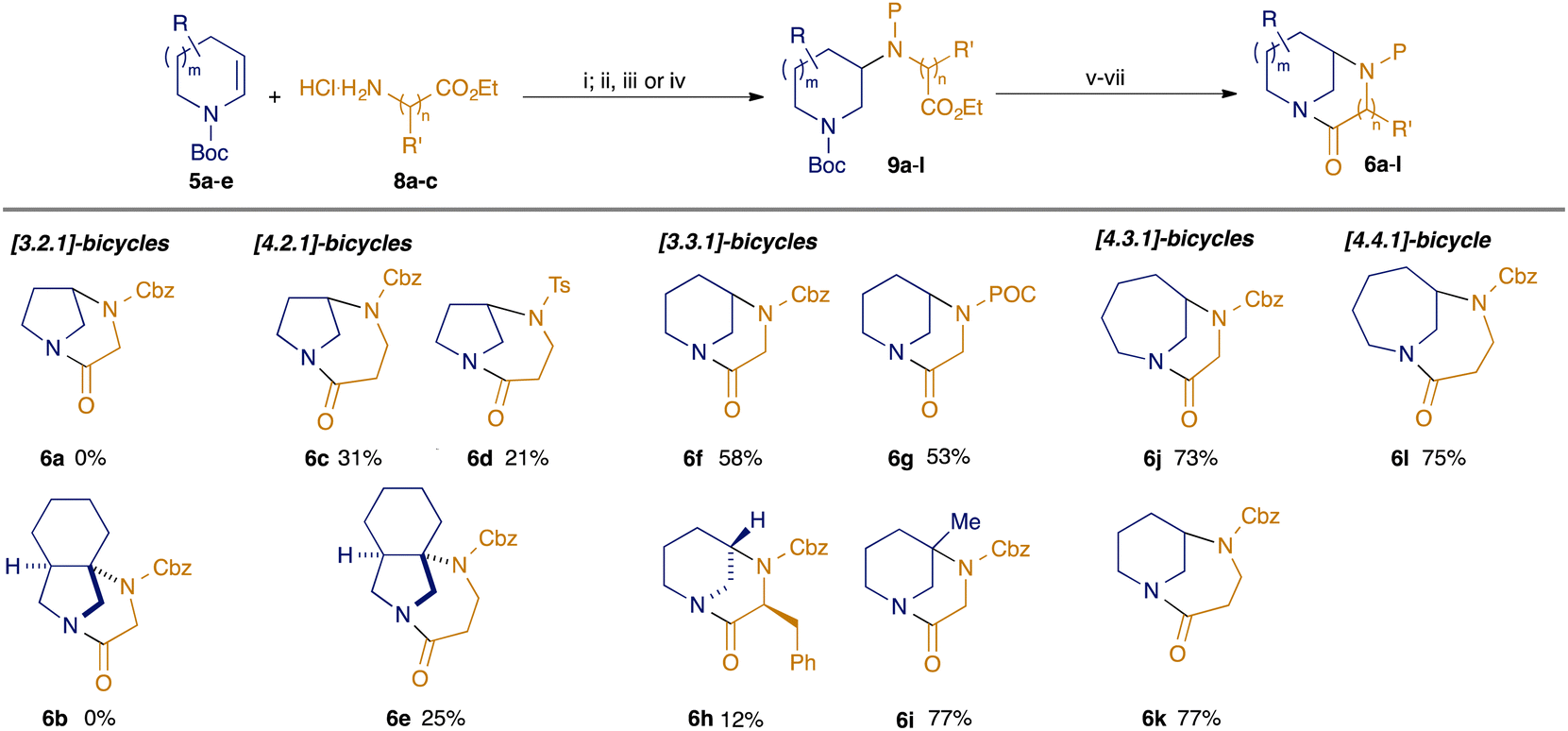 | ||
| Scheme 1 Modular synthesis of bicyclic twisted amides. (i) [Ir(dF(Me)ppy)2(dtbbpy)PF6] (2 mol%), TRIP thiol (50 mol%), toluene, blue LEDs; (ii) CbzCl, NaHCO3, DCM; (iii) TsCl, DIPEA, DMAP (5 mol%), DCM; (iv) POCCl (= propargyl chloroformate), NaHCO3, DCM; (v) NaOH, MeOH/H2O, 70 °C; (vi) 6M HCl, EtOAc; (vii) nBu2SnO, toluene, reflux. Yields shown are for the sequence from 9 to 6 (steps v–vii) only; for full details see ESI† | ||
Our studies commenced by exploiting our previously reported conditions13 for the photoredox-catalysed hydroamination of enecarbamates (Scheme 1). Hence, coupling of substrates 5a–e (prepared from the corresponding saturated amines through electrochemical oxidation and elimination15) with aminoesters 8a–c proceeded in generally good yield (see ESI† for full details). Protection of the resulting secondary amines variously as carbamates (P = Cbz, POC) or sulfonamides (P = Ts) enabled the preparation of a library of cyclisation precursors 9a–l. Conversion to the desired diazabicyclic scaffolds was effected by basic ester hydrolysis, removal of the Boc protecting group from the azacycle under acidic conditions, and finally cyclisation using Bu2SnO.12 Attempts to form the [3.2.1]-diazabicyclic products 6a,b derived from hydroamination of dihydropyrroles 5a,b with ethyl glycinate 8a failed at the cyclisation step, presumably owing to the high degree of strain in the products: we are unaware of any successful approaches to twisted amides with the [3.2.1]-bicyclic skeleton via amide bond formation. However, we were delighted to find that the use of ethyl aminopropionate 8b as the hydroamination partner led successfully to the formation of the diazabicyclo[4.2.1]nonanes 6c–e in moderate yield. Most notably, the formation of the unprecedented tricyclic system 6e, which contains a fully-substituted carbon, could not have been achieved through our previous reductive amination approach.12 Cyclisations of substrates based on hydroamination of Boc-tetrahydropyridine 5c with glycine ester 6a were also successful, in line with our previous construction of diazabicyclo[3.3.1]nonanes:12 the simple Cbz and POC-protected scaffolds 6f/g were returned in higher yield than the corresponding [4.2.1] products 6c–e, which may be indicative of the relative degrees of strain in the products (vide infra). The incorporation of backbone substituents on the coupling partners 5 and 8 was also probed: use of an α-substituted amino ester (ethyl phenylalaninate 8c) led to a mixture of diastereomers in the hydroamination step, only one of which cyclised to give a poor yield of the desired bicyclic product 6h (relative configuration determined by NOE studies, ESI†). Pleasingly, hydroamination using Boc 3-methyltetrahydropyridine 5d led ultimately to bridgehead-substituted bicycle 6i in a notably higher yield than for 6f, perhaps reflecting a lower energy penalty to access the reactive conformation for lactamisation: again, this substitution pattern would not be available through routes based upon reductive amination chemistry.
Larger ring systems containing diazabicyclo[4.3.1]decanes could be accessed either through hydroamination of Boc tetrahydropyridine 5c with ethyl aminopropionate 8b, or as an isomeric lactam through the coupling of the dehydroazepine 5e with ethyl glycinate 8a. Regardless of the location of the amide (in the six- or seven-membered ring in 6j and 6k respectively), the cyclisations occurred in good yield, as was also seen in the cyclisation to give the homologous diazabicyclo[4.4.1]undecane 6l.
Our new synthetic strategy had enabled us to prepare five distinct diazabicyclic bridged amides (and substituted/fused variants) varying systematically in the size of each ring, enabling interrogation of the impact on molecular properties (Fig. 2; data for ‘parent’ scaffolds shown, for all data see ESI†). The reduced contribution from resonance forms with CN character manifests itself in a greater degree of electron-deficiency at the carbonyl carbon, and hence higher 13C NMR chemical shifts3a (for example in the [4.n.1] series 6c/k/l, δC = 182.1, 176.8, 172.8 ppm for n = 2, 3, 4). Similarly, the greater overall contribution of the CO resonance form3a can be seen in the IR stretches (e.g. for the [4.n.1] series 6c/k/l, νmax = 1680, 1657, 1647 for n = 2, 3, 4).
 | ||
| Fig. 2 Influence of ring-size on structural and spectroscopic features. a two signals visible owing to Cbz rotamers, Δδ < 0.1 ppm; P = Cbz except for bvalues for P = Bn.12 | ||
Single crystal X-ray analysis of compound 6l also permits comparison with our previously reported structures of the N-Bn analogues of 6f/k.12 The significant pyramidalisation at nitrogen can be seen as a sum of the bond angles at that atom:16 while the [4.4.1] compound 6l shows near planarity (Σangles = 357.8° vs. ideal sp2 = 360°), the smaller homologues show significant pyramidalisation, with the diazabicyclo[3.3.1]nonane skeleton much closer to sp3 hybridisation (Σangles = 333.7° vs. ideal sp3 = 328.4°). Amide distortion can also be measured through the Winkler–Dunitz parameters:17 for example, the ‘twist angle’ τ (0° for a planar amide and 90° for a perpendicular arrangement) varies from 3.6–23.5° across the series (for additional parameters, see ESI†). Finally, as expected, the N–C bond shortens with increased conjugation of the nitrogen lone pair: the relatively long bond of 1.380 Å in the N–Bn analogue of 6f shortens to 1.355 Å in 6l (cf. 1.475 Å in the most-twisted amide 14a and 1.325 Å for N-methylpiperidone4b). We considered that the twist-induced destabilisation of (particularly) the smaller diazabicycles could make them competent acylating agents and hence they might have the potential to act as anti-microbial agents. Screening of a panel of compounds 6 was therefore carried out against Staphylococcus Aureus ATCC29213, with the strained tricyclic structure 6e showing activity at 32 μg mL−1 (see ESI† for full details).
The modular nature of our synthetic approach meant that it could be readily adapted to the synthesis of related twisted anilines (Scheme 2). Hence, photoredox-catalysed hydroamination of enecarbamates 5a, c and e with 2-bromobenzylamine 10a followed by suitable amine protection gave adducts 11a–d. Removal of the Boc-group was followed by intramolecular amine arylation under Buchwald–Hartwig conditions, giving the desired homologous diazabicycles 7a–d. We also attempted to investigate homologues in the diazacyclic ring, but although the desired cyclisation precursor 11e could be readily prepared by reaction of 5c with 2-(2-bromophenyl)ethan-1-amine 10b, the Buchwald–Hartwig cyclisation gave only a trace amount of the desired product 7e, reflecting the challenging nature of medium-ring formation.18
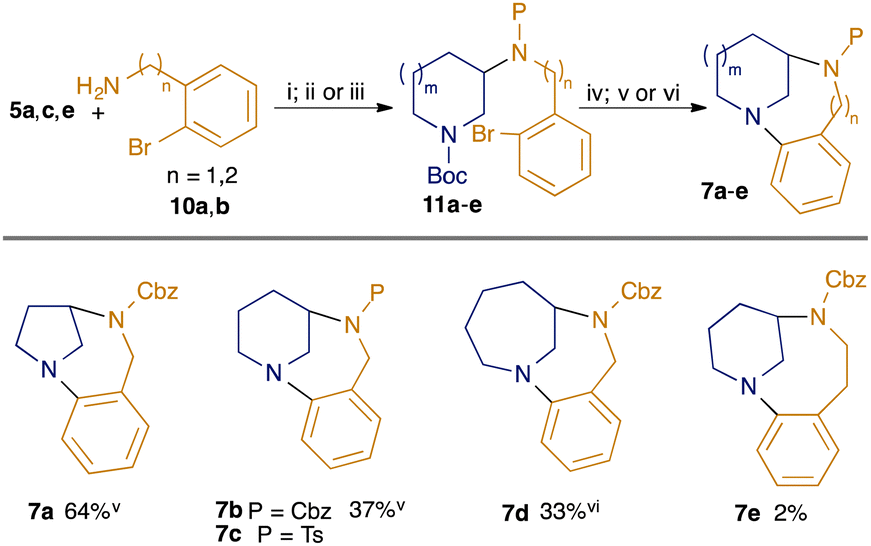 | ||
| Scheme 2 Modular synthesis of twisted bicyclic anilines. (i) [Ir(dF(Me)ppy)2(dtbbpy)PF6] (2 mol%), TRIP thiol (50 mol%), toluene, blue LEDs; (ii) CbzCl, NaHCO3, DCM; (iii) TsCl, DIPEA, DMAP (5 mol%), DCM; (iv) 6M HCl, EtOAc; (v) Pd2(dba)3 (4 mol%), BINAP (8 mol%), NaOtBu, toluene, reflux; (vi) Pd(OAc)2 (10 mol%), BINAP (20 mol%), Cs2CO3, toluene, reflux. Yields shown for steps from 11 to 7 (steps iv plus v/vi) only: for full details see ESI.† | ||
Products 7a–e were oils and so crystallographic structural data could not be obtained. However, the enforced non-planarity of the anilinic nitrogen with the aromatic ring may be inferred from the 13C NMR shift of the para-carbon to the nitrogen, as demonstrated by Zakrzewska.19 The values for 7a, b and d are shown in Fig. 3: the value of 118.1 ppm for the largest ring system 7d indicates a largely planar structure (cf. 115.1 ppm for N,N-dibutylaminobenzene and 118.9 ppm for N-phenylpiperidine19); the higher value of 121.4 for the dehomologous 7b suggests a departure from planarity, while the value of ca. 129 ppm for 7a indicates significant disruption of resonance (cf. 127.0 ppm for benzoquinuclidine 3,19 which features a completely orthogonal lone pair). As expected, the value for the relevant carbon in the benzodiazacene-containing 7e (119.2 ppm) indicates strong lone pair overlap.
 | ||
| Fig. 3 Influence of ring-size on 13C NMR shift and pKa of bicyclic anilines 7. a calculated values (DMSO solvation); b unambiguous assignment not possible. | ||
The different degrees of lone-pair overlap ought also to be reflected in the basicity of the nitrogen. Compounds 7 are poorly water soluble, and determination of pKa values in non-aqueous solvents is non-trivial. We recently developed an NMR-based assay using pH gradients in a single sample and chemical shift imaging to determine pKa values in DMSO.20,21 Using this method, a significant variation in pKa was observed: over 3 units separate the three compounds 7a,b,d in the homologous series, with the most strained variant 7a being the most basic as expected. This trend in relative basicity was also confirmed computationally in water using the Jaguar pKa prediction method.22,23 Inspection of the calculated structures23 (gas phase and solvated in DMSO or water; see ESI†) also revealed the expected trend in pyramidalisation at nitrogen (measured by sum of bond angles): 7a shows values close to that for ideal sp3-hybridisation, whereas 7d is much closer to that for sp2.
In summary, we have developed a modular approach to the synthesis of diazabicyclic amides and anilines, in which independent variation of the ring sizes allows interrogation of the effect on molecular structures and properties. The ability to tune molecular properties (e.g. pKa) by variation of scaffold size rather than substituents or functional groups may find application in the design of functional organic molecules.
We thank EPSRC (EP/N025652/1)/Redbrick Molecular (studentship to AH), AstraZeneca (Grant 10045297)/University of Liverpool (financial support to KB), and Schrödinger for a trial license of Small Molecule Drug Discovery suite. We thank Julian Chesti (Leeds) for the anti-microbial assay. Data associated with this paper are openly available in the ESI.† For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising from this submission.
Conflicts of interest
Redbrick Molecular marketed diverse building blocks for applications in drug discovery.Notes and references
- (a) L. Pauling, The nature of the chemical bond, Cornell University Press, Ithaca, 1940 Search PubMed; (b) L. Pauling, R. B. Corey and H. R. Branson, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 205–211 CrossRef CAS PubMed; (c) R. B. Corey and L. Pauling, Proc. R. Soc. B, 1953, 141, 10–20 CAS.
- A. S. Edison, Nat. Struct. Biol., 2001, 8, 201–202 CrossRef CAS PubMed.
- (a) M. Szostak and J. Aubé, Chem. Rev., 2013, 133, 5701–5765 CrossRef PubMed; (b) C. Liu and M. Szostak, Chem. – Eur. J., 2017, 23, 7157–7173 CrossRef CAS PubMed; (c) G. Meng, J. Zhang and M. Szostak, Chem. Rev., 2021, 121, 12746–12783 CrossRef CAS PubMed.
- (a) A. J. Kirby, I. V. Komarov, P. D. Wothers and N. Feeder, Angew. Chem., Int. Ed., 1998, 37, 785–786 CrossRef CAS; (b) A. J. Kirby, I. V. Komarov, K. Kowski and P. Rademacher, J. Chem. Soc., Perkin Trans. 2, 1999, 1313–1316 RSC; (c) I. V. Komarov, S. Yanik, A. Y. Ishchenko, J. E. Davies, J. M. Goodman and A. J. Kirby, J. Am. Chem. Soc., 2015, 137, 926–930 CrossRef CAS PubMed.
- (a) K. Tani and B. M. Stoltz, Nature, 2006, 441, 731–734 CrossRef CAS PubMed; (b) T. Ly, M. Krout, D. K. Pham, K. Tani, B. M. Stoltz and R. R. Julian, J. Am. Chem. Soc., 2007, 129, 1864–1865 CrossRef CAS PubMed.
- R. Szostak, J. Aubé and M. Szostak, Chem. Commun., 2015, 51, 6395–6398 RSC.
- B. M. Wepster, Recueil, 1952, 71, 1159–1170 CrossRef CAS.
- (a) J. Tröger, J. Prakt. Chem., 1887, 36, 225–245 CrossRef; (b) Ö. V. Rúnarsson, J. Artacho and K. Wärnmark, Eur. J. Org. Chem., 2012, 7015–7041 CrossRef.
- A. J. Hoefnagel, M. A. Hoefnagel and B. M. Wepster, J. Org. Chem., 1981, 46, 4209–4211 CrossRef CAS.
- R. P. Duke, R. A. Y. Jones, A. R. Katritzky, E. E. Mikhlina, A. D. Yanina, L. M. Alekseeva, K. F. Turchin, Y. N. Sheinker and L. N. Yakhontov, Tetrahedron Lett., 1970, 11, 1809–1812 CrossRef.
- (a) G. Köhler, G. Grabner and K. Rotkiewicz, Chem. Phys., 1993, 173, 275–290 CrossRef; (b) T. Okada, M. Uesugi, G. Köhler, K. Rechthaler, K. Rotkiewicz and W. Rettig, et al. , Chem. Phys., 1999, 241, 327–337 CrossRef CAS.
- H. Hassan, S. P. Marsden and A. Nelson, Bioorg. Med. Chem., 2018, 26, 3030–3033 CrossRef CAS PubMed.
- D. Francis, A. Nelson and S. P. Marsden, Chem. – Eur. J., 2020, 26, 14861–14865 CrossRef CAS PubMed.
- A. J. Musacchio, B. C. Lainhart, X. Zhang, S. G. Naguib, T. C. Sherwood and R. R. Knowles, Science, 2017, 355, 727–730 CrossRef CAS PubMed.
- A. F. Trindade, E. L. Faulkner, A. G. Leach, A. Nelson and S. P. Marsden, Chem. Commun., 2020, 56, 8802–8805 RSC.
- Y. Otani, O. Nagae, Y. Naruse, S. Inagaki, M. Ohno, K. Yamaguchi, G. Yamamoto, M. Uchiyama and T. Ohwada, J. Am. Chem. Soc., 2003, 125, 15191–15199 CrossRef CAS PubMed.
- F. K. Winkler and J. D. Dunitz, J. Mol. Biol., 1971, 59, 169–182 CrossRef CAS PubMed.
- For examples of azocine formation by Pd-cat. amination: (a) A. Neogi, T. P. Majhi, R. Mukhopadhyay and P. Chattopadhyay, J. Org. Chem., 2006, 71, 3291–3294 CrossRef CAS PubMed; (b) N. D. Adhikary and P. Chattopadhyay, Eur. J. Org. Chem., 2010, 1754–1762 CrossRef; (c) S. Mitra, T. S. Banerjee, S. K. Hota, D. Bhattacharya, S. Das and P. Chattopadhyay, Eur. J. Med. Chem., 2011, 46, 1713–1720 CrossRef CAS PubMed For Cu-mediated variants, see e.g.: ; (d) J. L. Kenwright, W. R. J. D. Galloway, D. T. Blackwell, A. Isidro-Llobet, J. Hodgkinson, L. Wortmann, S. D. Bowden, M. Welch and D. R. Spring, Chem. – Eur. J., 2011, 17, 2981–2986 CrossRef CAS PubMed.
- A. Zakrewska, R. Gawinecki, E. Kolehmainen and B. Osmialowski, Int. J. Mol. Sci., 2005, 6, 52–62 CrossRef.
- G. Schenck, K. Baj, J. A. Iggo and M. Wallace, Anal. Chem., 2022, 94, 8115–8119 CrossRef CAS PubMed.
- M. Wallace, D. J. Adams and J. A. Iggo, Anal. Chem., 2018, 90, 4160–4166 CrossRef CAS PubMed.
- (a) A. D. Bochevarov, M. A. Watson, J. R. Greenwood and D. M. Philipp, J. Chem. Theory Comput., 2016, 12, 6001–6019 CrossRef CAS PubMed; (b) H. S. Yu, M. A. Watson and A. D. Bochevarov, J. Chem. Inf. Model., 2018, 58, 271–286 CrossRef CAS PubMed; (c) J. J. Klicić, R. A. Friesner, S.-Y. Liu and W. C. Guida, J. Phys. Chem. A, 2002, 106, 1327–1335 CrossRef; (d) Schrödinger Release 2023-1: Jaguar pKa, Schrödinger, LLC, New York, NY, 2021 Search PubMed.
- (a) A. D. Bochevarov, E. Harder, T. F. Hughes, J. R. Greenwood, D. A. Braden, D. M. Philipp, D. Rinaldo, M. D. Halls, J. Zhang and R. A. Friesner, Int. J. Quantum Chem., 2013, 113, 2110–2142 CrossRef CAS; (b) Schrödinger Release 2023-1: Maestro, Schrödinger, LLC, New York, NY, 2021 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2192693. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc01470c |
| This journal is © The Royal Society of Chemistry 2023 |