
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective alkylation of mandelic acid to diarylacetic acids over a commercial zeolite†
Samuel G.
Meacham
and
Russell A.
Taylor
 *
*
Department of Chemistry, Durham University, South Road, Durham DH1 3LE, UK. E-mail: russell.taylor@durham.ac.uk
First published on 6th July 2023
Abstract
A commercial zeolite is shown to be a highly effective heterogeneous catalyst for the Friedel–Crafts alkyation of mandelic acid with aromatic substrates. The reaction yields mixed diarylacetic acids in one step avoiding the need for inert atmosphere techniques or superacids. The observed reaction pathways are zeolite framework dependent with only the FAU framework giving very high selectivity to the mixed diarylacetic acids.
The Friedel–Crafts (FC) alkylation reaction is catalysed by a wide variety of strong Lewis acids (e.g. AlCl3) and Brønsted acids (e.g. H2SO4), but very commonly stoichiometric, or super-stoichiometric, amounts of these acids are utilised.1,2 The traditional use of toxic alkylhalide substrates as masked electrophiles also leads to problems with the formation of HX by-products, such as salt formation. As such, the development of improved Friedel–Crafts alkylation procedures for the formation of C–C bonds remains a highly active area of chemical research.3–5 The use of so-called “π-activated” alcohols has proven to be a successful way to use alcohols as electrophiles (rather than toxic alkylhalides) but the great number of reports in the field utilise transition metal catalysts or metal salts that can be de-activated by the water co-produced and/or are difficult to recycle.3,6 The use of heterogeneous catalysts for Friedel–Crafts alkylation reactions with alcohols has the potential to overcome the aforementioned limitations, with heteropolyacids and zeolites showing utility in this general transformation.7–12
We herein report that a commercial zeolite, H-Y-30 (CBV760 from Zeolyst), selectively converts mandelic acid to diarylacetic acids through an FC alkylation reaction (hydroxyalkylation) in aromatic solvents. Diarylacetic acid moieties are important scaffolds in active pharmaceutical ingredients13–17 but simple synthetic routes do not exist at present. Homogeneous routes to mixed diarylacetates have been reported from monoaryl precursors, but many of these routes are expensive (e.g. Pd plus ligand18,19), atom inefficient (e.g. stoichiometric metal salts20,21 or halogenated substrates18–21) and/or hazardous to implement (diazo-precursors22–24).
On the other hand, diarylacetic acids can be formed with heterogeneous, strong Brønsted acid catalysts. Super-stoichiometric triflic acid supported on PVP can catalyse the formation of diarylacetic acids directly from glycolic acid and aromatics in one step, but the catalyst is air sensitive and requires handling of hazardous, super acid: triflic acid.25 Similarly, H2SO4 supported on silica (in glacial acetic acid) can catalyse the same reaction when using electron rich aromatics, but was unable to convert mandelic acid to a diarylacetic acid.26
During the course of our investigations into the zeolite catalysed reactions of mandelic acid, we have discovered that mixed diarylacetic acids can be formed from mandelic acid and aromatic substrates using a commercial acidic zeolite catalyst, without the need for inert atmosphere techniques. Initial catalyst screening results with p-toluenesulfonic acid (pTSA) and a variety of acidic zeolite catalysts are shown in Fig. 1.
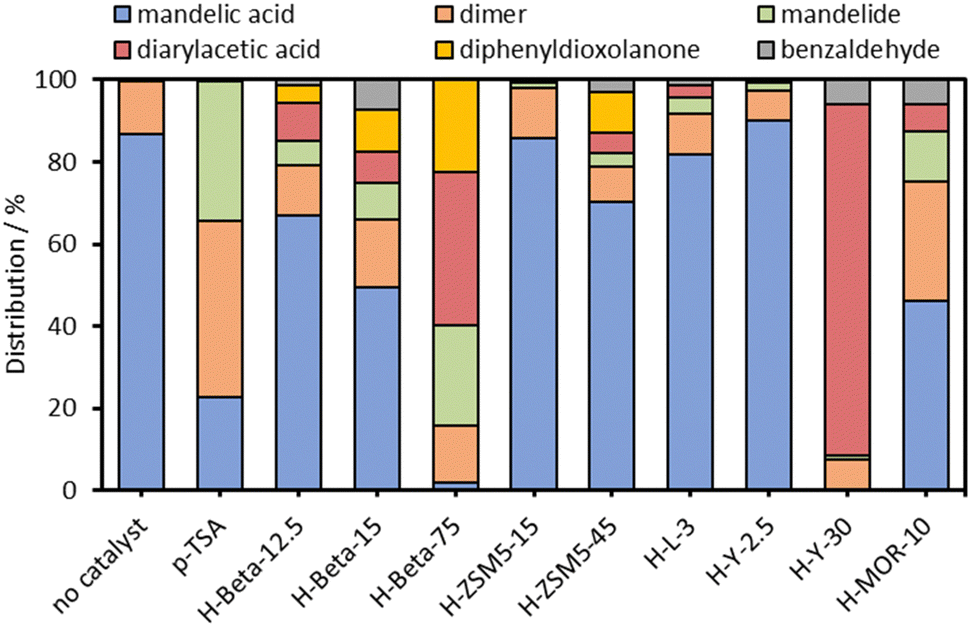 | ||
| Fig. 1 Effect of zeolite catalyst framework and Si/Al ratio on product distribution. Conditions: 0.5 g mandelic acid in 50 ml mixed xylenes, 3 mol% H+ (pTSA) or 3 mol% Al (zeolites), reflux with Dean Stark trap overnight (ca. 20 hours), stirring rate = 500 rpm. | ||
Using mixed xylenes as substrate and solvent, the overnight reactions were carried out using a phase separator to remove water from the reaction flask (Fig. S1, ESI†). Zeolite catalyst loadings were adjusted to give 3 mol% Al (relative to mandelic acid). Fig. 1 shows that when using a homogeneous catalyst (pTSA) only the linear dimer and cylic dimer, mandelide, were produced at ∼80% conversion. The formation of mandelide using pTSA as catalyst is known27,28 (mixed xylenes, 1 day, 25% or 3 days, 57% yield) and was reproduced under our conditions. However, when using heterogeneous, acidic zeolite catalysts additional products were observed alongside the expected linear dimer and mandelide (Scheme 1). These additional products were: (1) diarylacetic acids from FC alkylations, (2) benzaldehyde, and (3) 2,5-diphenyl-1,3-dioxolan-4-one from the reaction of benzaldehyde with mandelic acid.
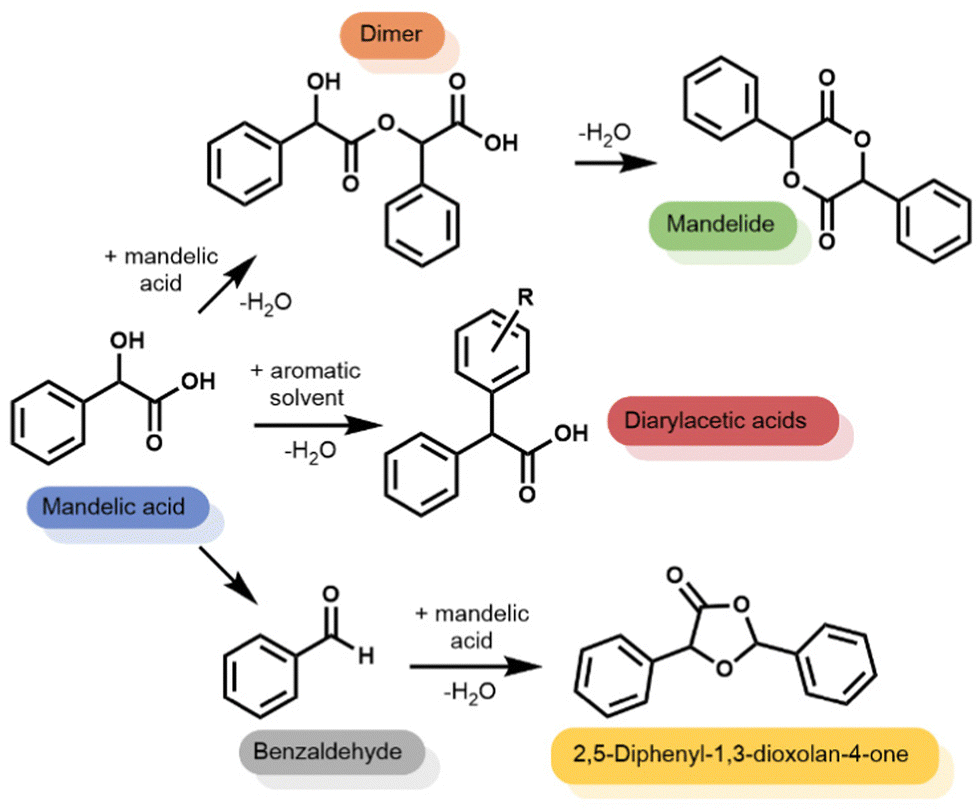 | ||
| Scheme 1 Range of products observed in zeolite-catalysed reactions of mandelic acid. | ||
From Fig. 1 it is clear that the zeolite framework and Si/Al ratio have a marked effect on conversion and the observed product distribution. Zeolites H-Beta-75 and H-Y-30 were the most active for mandelic acid conversion under the reaction conditions. For H-Beta, three different Si/Al ratios were tested and we surprisingly observed that the conversion increased from approximately 35% to 98% upon increasing the Si/Al from 12.5 to 75, despite the Al loading remaining constant. In the case of zeolite H-Y-30, very high selectivity for the FC alkylation reaction was observed. The medium pore ZSM-5 zeolites showed lower conversion than the large pore H-Beta, H-Y and H-MOR zeolites, which may be due to the reactant being too large to enter the medium micropore system of ZSM-5.
In order to further understand the reaction pathways occurring with the most active materials, H-Y-30 and H-Beta-75, a series of reaction time profile experiments were carried out, the results of which are shown in Fig. 2. In the case of H-Beta-75, all reaction pathways (shown in Scheme 1) occur simultaneously. At short reaction times, the linear dimer of mandelic acid and the FC alkylation product are the major components. At reaction times longer than 1 hour, the linear dimer concentration decreases as it is converted to the cyclic dimer mandelide. Benzaldehyde is also observed at all reaction times, with the subsequent dioxolanone product only forming at reaction times greater than 1 hour. Between 2 to 3.5 hours, the predominant reaction occurring is the FC Alkylation. For H-Y-30 the major product is the FC alkylation product at all time points measured.
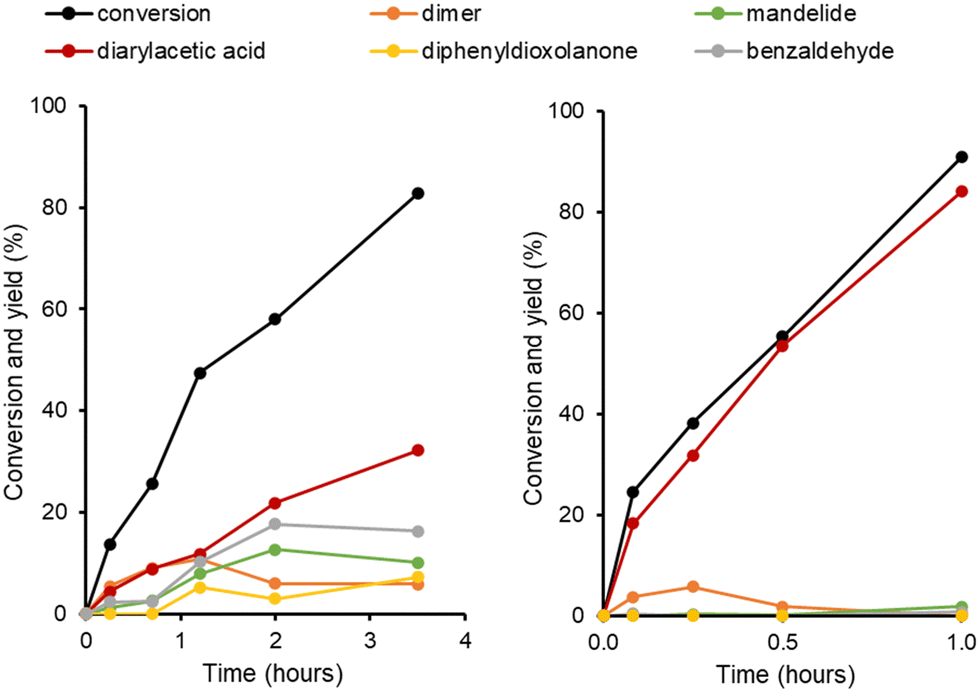 | ||
| Fig. 2 Effect of reaction time on product distribution for H-Beta-75 (left) and H-Y-30 (right) zeolite-catalysed reaction of mandelic acid. Solvent = mixed xylenes. | ||
The FC alkylation of mandelic acid catalysed by H-Y-30 was explored further using a range of aromatic solvents (o-, m-, p-xylene, toluene and chlorobenzene). Initial experiments (reflux, phase separator, 1 hour) show that the FC alkylation product dominates across all solvents (Fig. S15, ESI†), but conversion varied from 17% (toluene) up to 92% (o-xylene), and selectivity for the FC product varied from 68% (chlorobenzene) to 97% (o-xylene). As this variation in selectivity could be due to the different reflux temperatures (and thus variation of selectivity at different conversion levels after 1 hour) or aromatic substituent effects, time profiles were also recorded (Fig. 3A) in order to study product selectivity at near iso-conversion (Fig. 3B). The selectivity to the FC alkylation product still varies at near iso-conversion (Fig. 3B), indicating that the aromatic substituents do cause directing effects and modulation of ring reactivity. Such effects are consistent with carbocation chemistry, suggesting carbocation intermediates may be involved. Screening H-Beta-75 across the same range of reaction solvents still resulted in a broad range of products; notably, in the absence of aromatic solvents, mandelide could be formed as the dominant product over H-Beta-75 (Fig. S16, ESI†). An additional iso-conversion study comparing H-Beta-75, H-Y-30, H-ZSM5-45 and H-MOR-97 show that the high selectivity to diarylacetic acids is framework selective (Fig. S17, ESI†).
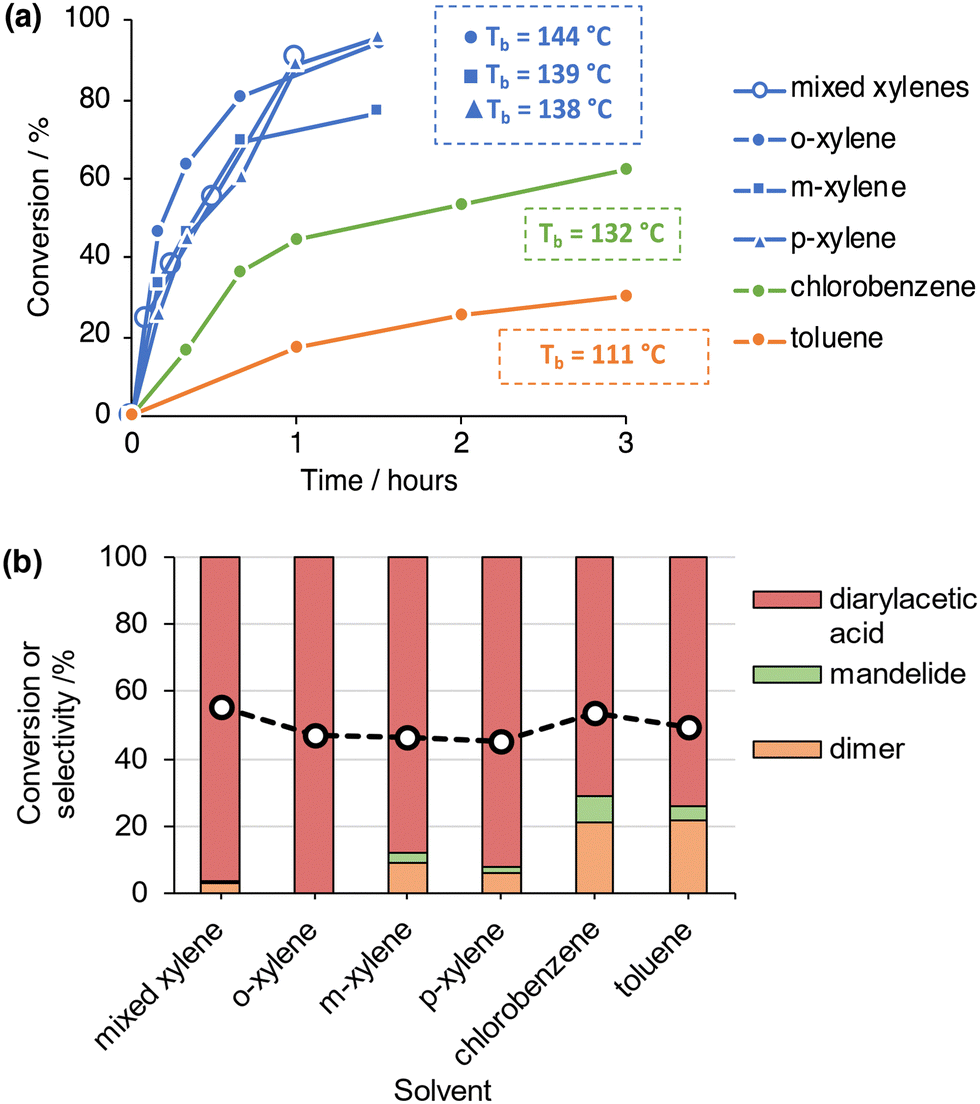 | ||
| Fig. 3 (a) Effect of reaction time on mandelic acid conversion in various aromatic solvents. (b) Selectivity and conversion to arylation products catalysed by H-Y-30 zeolite. Reactions were run for various time under reflux at the boiling point of each solvent to obtain 45–55% conversion. Aside from p-xylene, products are obtained as a mixture of positional isomers, with the para isomer the major product. | ||
These observations show that the product of mandelic acid conversion with aromatics by acidic zeolite catalysts can be tuned by controlling the microenvironment surrounding the active site. Homogenous pTSA yields condensation products of dehydration, while zeolites can promote an FC alkylation pathway that is typical of superacids,25,26 but do so under non-superacid conditions The yield of the FC reaction product is highly dependent on the zeolite framework, indicating that the necessary reaction intermediates for FC alkylation of mandelic acid are stabilised by the FAU framework. In addition, the observation that H2SO4 supported on silica (in glacial acetic acid) is unable to convert mandelic acid to a diarylacetic acid in aromatic solvents26 supports the importance of the zeolite framework in modulating the reaction outcome. The observation of multiple reaction pathways, catalysed by zeolites, for mandelic acid conversion under these reaction conditions is in contrast to a recent report which showed that H-Y-2.6, H-Beta-12.5 and H-Beta-75 were effective catalysts for the production of lactide from lactic acid (in xylenes, under analogous conditions).29 The only by-products observed in this reported system were linear oligomers of lactic acid, in contrast to our findings. Furthermore, surfactant-templated zeolite Y has been successfully applied for the FC alkylation of indole with “π-activated” alcohols.11 Indole is, however, an electron-rich nucleophile in FC reactions with a nucleophilicity parameter 9 orders of magnitude greater than toluene and is therefore easily alkylated in FC alkylation reactions.30,31 In addition, “π-activated” alcohols bearing electron withdrawing groups are notoriously difficult FC electrophiles,32 all of which highlights the remarkable selectivity exhibited with H-Y-30 for the reported FC alkylation reactions reported here.
To further consider the differences in product selectivity observed across the range of zeolite catalysts tested, the size of some of the key products formed in these experiments were modelled using an approach that has been successfully reported recently.33,34 Molecular sizes were approximated by first carrying out an energy minimisation of the molecular structures followed by calculating the minimum projection area (from van der Waals radii) as detailed in the ESI† (Section S3). The diameter of mandelic acid (Fig. 4) was found to be approximately 7.1 Å, greater than the largest pore width of both zeolites ZSM-5 (5.7 Å) and H-Beta (6.6 Å), but slightly smaller than that of H-Y (7.4 Å).35 Mandelide has a similar diameter of 7.3 Å. The largest of all the products are the diarylacetic acids, at 8.4 Å for the product of p-xylene and mandelic acid. This is larger than the nominal pore widths of all zeolites tested in this work but could be included within the supercage of zeolite Y (11.2 Å). Hence mandelic acid is very likely excluded from accessing the internal pore system of medium pore (10-membered ring) H-ZSM-5, resulting in low conversion. However, for the large (12-membered ring) zeolites such as Beta, Y and MOR, conversion during screening was observed to be more significant in general (Fig. 1) suggesting improved active site accessibility. For the same zeolite framework, conversion was shown to increase as the Si/Al ratio increases (Fig. 1). This is exemplified for zeolite H-Beta where the conversion increases across three Si/Al ratios (12.5, 15 and 75) but the product distribution remains constant. Likewise, for zeolite H-Y, conversion increases across H-Y-2.5, H-Y-15 and H-Y-30 (5%, 38% and 90% respectively, (1 hour, p-xylene, reflux, Fig. S18, ESI†). The formation of benzaldehyde and dioxolanone is not observed for any H-Y catalysts (Fig. S18, ESI†). It has been well established that the high silica zeolite Y materials from Zeolyst (CBV series) are formed by steaming and acid washing and result in hierarchical materials containing mesopores.36,37 The fact that high silica forms of both Beta and Y, which all have higher mesoporosity (ESI,† Section S4) and give rise to the highest conversion, indicate that the presence of mesopores are critical for high catalytic activity.
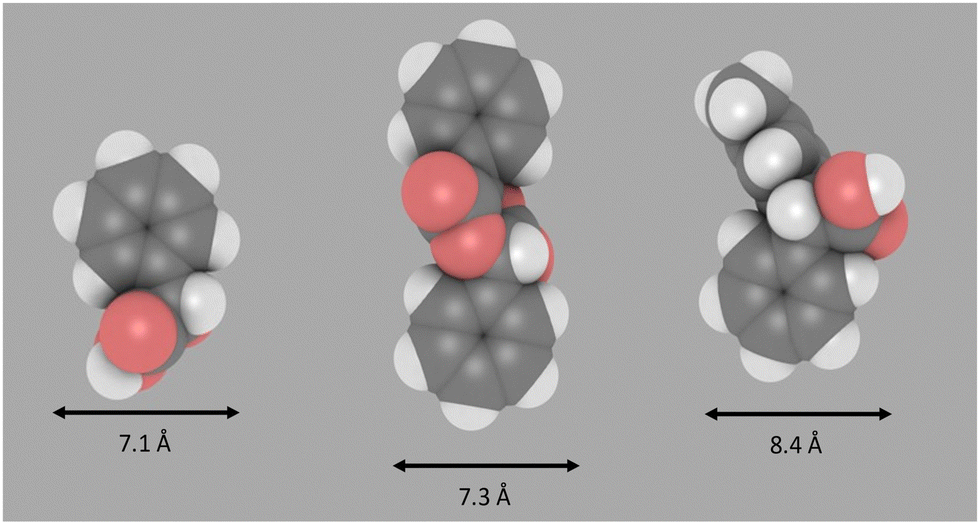 | ||
| Fig. 4 Approximate diameter perpendicular to long axis of the molecule. Image produced using iRASPA. Left: Mandelic acid, middle: mandelide, right: Alpha arylation product (solvent = p-xylene). | ||
Whilst the true origin of the observed selectivity for FC alkylation products is beyond the scope of this work, the following observations are suggestive. We have calculated that diarylacetic acids should be too large to exit (or enter) the micropore system of any of the frameworks tested, likely excluding the traditional modes of shape selectivity (reactant, product and transition) that are well established in zeolite catalysis.38–40 Counter intuitively, an inverse relationship of conversion with acid site density is observed for Beta and FAU frameworks, but this is coupled to the hierarchical nature of the high silica forms of FAU and Beta catalysts tested here, which indicates that hierarchical structures are critical to conversion. In addition, the very high selectivity for FC alkylation over high silica, hierarchical H-Y-30, which is not observed over other frameworks or with homogeneous catalysts, indicates that the (heirachical) FAU framework is essential. This suggests that confinement41 or the nest effect40 (i.e. surface exposed, partial super cages) may be responsible for our observations. We are continuing our investigations to probe this facile procedure and determine the origins of the observed selectivity.
In conclusion, we have shown that the observed product selectivity in the reaction of mandelic acid in aromatic solvents is controlled by the zeolite frameworks, and diarylacetic acids can be formed selectivity over the FAU framework. The findings provide a simple and green approach to this important organic moiety, without the need for super-stoichiometric acid, or corrosive and/or inert conditions.
R. A. T. thanks the EPSRC for generous funding of an EPSRC Manufacturing Fellowship, EP/R01213X/1. S. M. was supported by the EPSRC CDT in Soft and Functional Interfaces, EP/L015536/1.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Rueping and B. J. Nachtsheim, Beilstein J. Org. Chem., 2010, 6, 6 CrossRef PubMed.
- M. M. Heravi, V. Zadsirjan, P. Saedi and T. Momeni, RSC Adv., 2018, 8, 40061–40163 RSC.
- R. Kumar and E. V. Van der Eycken, Chem. Soc. Rev., 2013, 42, 1121–1146 RSC.
- S. Zhang, M. Vayer, F. Noël, V. D. Vuković, A. Golushko, N. Rezajooei, C. N. Rowley, D. Lebœuf and J. Moran, Chemistry, 2021, 7, 3425–3441 CrossRef CAS.
- S. Estopiñá-Durán and J. E. Taylor, Chem. – Eur. J., 2021, 27, 106–120 CrossRef PubMed.
- M. Bandini and M. Tragni, Org. Biomol. Chem., 2009, 7, 1501–1507 RSC.
- K.-i Shimizu and A. Satsuma, Energy Environ. Sci., 2011, 4, 3140–3153 RSC.
- Y. Sun and R. Prins, Appl. Catal., A, 2008, 336, 11–16 CrossRef CAS.
- K. Masuda, Y. Okamoto, S. Y. Onozawa, N. Koumura and S. Kobayashi, RSC Adv., 2021, 11, 24424–24428 RSC.
- Y. N. Nayak, N. Swarnagowri, Y. F. Nadaf, N. S. Shetty and S. L. Gaonkar, Lett. Org. Chem., 2020, 17, 491–506 CrossRef CAS.
- N. Linares, F. G. Cirujano, D. E. De Vos and J. García-Martínez, Chem. Commun., 2019, 55, 12869–12872 RSC.
- J. R. Cabrero-Antonino, A. Leyva-Pérez and A. Corma, Angew. Chem., Int. Ed., 2015, 54, 5658–5661 CrossRef CAS PubMed.
- D. M. Brown, B. O. Hughes, C. D. Marsden, J. C. Meadows and B. Spicer, Br. J. Pharmacol., 1973, 47, 476–486 CrossRef CAS PubMed.
- M. G. Palfreyman, E. S. Palfreyman and M. S. G. Clark, Eur. J. Pharmacol., 1974, 28, 379–383 CrossRef CAS PubMed.
- B. E. Maryanoff, S. O. Nortey and J. F. Gardocki, J. Med. Chem., 1984, 27, 1067–1071 CrossRef CAS PubMed.
- I. Roufos, S. J. Hays, D. J. Dooley, R. D. Schwarz, G. W. Campbell and A. W. Probert, Jr., J. Med. Chem., 1994, 37, 268–274 CrossRef CAS PubMed.
- I. Roufos, S. Hays and R. D. Schwarz, J. Med. Chem., 1996, 39, 1514–1520 CrossRef CAS PubMed.
- B. Song, T. Himmler and L. J. Gooßen, Adv. Synth. Catal., 2011, 353, 1688–1694 CrossRef CAS.
- W. A. Moradi and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7996–8002 CrossRef CAS PubMed.
- Y. Kim, Y. S. Choi, S. K. Hong and Y. S. Park, Org. Biomol. Chem., 2019, 17, 4554–4563 RSC.
- P.-S. Lai, J. A. Dubland, M. G. Sarwar, M. G. Chudzinski and M. S. Taylor, Tetrahedron, 2011, 67, 7586–7592 CrossRef CAS.
- S. Hu, J. Wu, Z. Lu, J. Wang, Y. Tao, M. Jiang and F. Chen, ChemCatChem, 2021, 13, 2559–2563 CrossRef CAS.
- Y. Xi, Y. Su, Z. Yu, B. Dong, E. J. McClain, Y. Lan and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 9817–9821 CrossRef CAS PubMed.
- B. Wang, I. G. Howard, J. W. Pope, E. D. Conte and Y. Deng, Chem. Sci., 2019, 10, 7958–7963 RSC.
- G. K. S. Prakash, F. Paknia, A. Kulkarni, A. Narayanan, F. Wang, G. Rasul, T. Mathew and G. A. Olah, J. Fluorine Chem., 2015, 171, 102–112 CrossRef CAS.
- D. L. Moore, A. E. Denton, R. M. Kohinke, B. R. Craig and W. E. Brenzovich, Synth. Commun., 2016, 46, 604–612 CrossRef CAS.
- T. Q. Liu, T. L. Simmons, D. A. Bohnsack, M. E. Mackay, M. R. Smith and G. L. Baker, Macromolecules, 2007, 40, 6040–6047 CrossRef CAS.
- G. J. Graulus, N. Van Herck, K. Van Hecke, G. Van Driessche, B. Devreese, H. Thienpont, H. Ottevaere, S. Van Vlierberghe and P. Dubruel, React. Funct. Polym., 2018, 128, 16–23 CrossRef CAS.
- M. Dusselier, P. Van Wouwe, A. Dewaele, P. A. Jacobs and B. F. Sels, Science, 2015, 349, 78–80 CrossRef CAS PubMed.
- H. Mayr, B. Kempf and A. R. Ofial, Acc. Chem. Res., 2003, 36, 66–77 CrossRef CAS PubMed.
- S. Lakhdar, M. Westermaier, F. Terrier, R. Goumont, T. Boubaker, A. R. Ofial and H. Mayr, J. Org. Chem., 2006, 71, 9088–9095 CrossRef CAS PubMed.
- V. D. Vuković, E. Richmond, E. Wolf and J. Moran, Angew. Chem., Int. Ed., 2017, 56, 3085–3089 CrossRef PubMed.
- T. C. Keller, S. Isabettini, D. Verboekend, E. G. Rodrigues and J. Pérez-Ramírez, Chem. Sci., 2014, 5, 677–684 RSC.
- F. C. Hendriks, D. Valencia, P. C. A. Bruijnincx and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2017, 19, 1857–1867 RSC.
- C. Baerlocher and L. McCusker, Database of Zeolite Structures, https://www.iza-structure.org/databases/, (accessed 23rd March 2023).
- K. P. de Jong, J. Zečević, H. Friedrich, P. E. de Jongh, M. Bulut, S. van Donk, R. Kenmogne, A. Finiels, V. Hulea and F. Fajula, Angew. Chem., Int. Ed., 2010, 49, 10074–10078 CrossRef CAS PubMed.
- J. Kenvin, S. Mitchell, M. Sterling, R. Warringham, T. C. Keller, P. Crivelli, J. Jagiello and J. Perez-Ramirez, Adv. Funct. Mater., 2016, 26, 5621–5630 CrossRef CAS.
- S. M. Csicsery, Zeolites, 1984, 4, 202–213 CrossRef CAS.
- M. E. Davis, Ind. Eng. Chem. Res., 1991, 30, 1675–1683 CrossRef CAS.
- T. F. Degnan, J. Catal., 2003, 216, 32–46 CrossRef CAS.
- R. Gounder and E. Iglesia, Chem. Commun., 2013, 49, 3491–3509 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc01444d |
| This journal is © The Royal Society of Chemistry 2023 |