
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Localised degradation within sulfide-based all-solid-state electrodes visualised by Raman mapping†
Jungwoo
Lim
 ab,
Yundong
Zhou‡
ab,
Rory H.
Powell
ab,
Tugce
Ates
cd,
Stefano
Passerini
cde and
Laurence J.
Hardwick
*ab
ab,
Yundong
Zhou‡
ab,
Rory H.
Powell
ab,
Tugce
Ates
cd,
Stefano
Passerini
cde and
Laurence J.
Hardwick
*ab
aStephenson Institute for Renewable Energy, Department of Chemistry, University of Liverpool, Liverpool L69 7ZF, UK. E-mail: hardwick@liverpool.ac.uk
bThe Faraday Institution, Harwell Campus, Didcot, OX11 0RA, UK
cHelmholtz Institute Ulm (HIU), Helmholtzstrasse 11, Ulm 89081, Germany
dKarlsruhe Institute of Technology (KIT), P.O. Box 3640, Karlsruhe 76021, Germany
eChemistry Department, Sapienza University of Rome, Piazzale A. Moro 5, Rome 00185, Italy
First published on 1st June 2023
Abstract
The distribution of degradation products, before and after cycling, within common sulfide-based solid electrolytes (β-Li3PS4, Li6PS5Cl and Li10GeP2S12) was mapped using Raman microscopy. All composite electrodes displayed the appearance of side reaction products after the initial charge-discharge cycle, located at the site of a LiNi0.6Mn0.2Co0.2O2 particle.
The development of sulfide-based lithium superionic conductors (>10−4 S cm−1) has addressed the challenge of low ionic conductivity, spurring the development of all-solid-state batteries (ASSB).1,2 Despite the suitable ionic conductivity of sulfide-based materials, they can be challenging to handle due to interfacial instability against active materials (electrodes and conductive binder) within the cell. The limited electrochemical window of sulfide solid electrolytes (of ca. 2–3 V vs. Li/Li+) can trigger chemical and electrochemical decomposition within the cell that leads to limited cell life.3 Previously, solid electrolyte decomposition has been analysed ex situ from recovered pellets or powders after reaction by X-ray photoelectron spectroscopy, powder X-ray diffraction and time-of-flight secondary ion mass spectrometry.4–6 Furthermore, Raman microscopy has been widely used for analysing ASSB electrodes, both in situ and ex situ, due to the ability to spatially map the chemical composition down to micron scale resolution. Raman mapping enables facile analysis, as well as direct chemical visualisation of the evolution of interfaces within practical electrode composites, after or during electrochemical treatment.7–9
To study the interfacial behaviour within more representative cell types, the scalable method of tape-casting (wet slurry fabrication) was adopted for electrode stack preparation10,11 and analysed by ex situ Raman microscopy. Previously, Raman spectroscopy has been used to map the charge heterogeneity of positive electrodes7 and in situ Raman analysis has highlighted the decomposition behaviour of Li6PS5Cl at the interface of both LiCoO2 and lithium metal.9
LiNi0.6Mn0.2Co0.2O2 was charged and discharged against lithium metal using one of three sulfide-based solid electrolytes (β-Li3PS4, Li6PS5Cl or Li10GeP2S12) from 3 to 4.2 V (vs. Li/Li+). Charging capacity among the cells showed slight variation between them: 159 mA h g−1 for β-Li3PS4, 172 mA h g−1 for Li6PS5Cl and 183 mA h g−1 for Li10GeP2S12 (Fig. 1). The variability derives from small differences in ionic conductivities and contact resistances between the three studied solid electrolytes and the active material LiNi0.6Mn0.2Co0.2O2. Li10GeP2S12 has the highest reported ionic conductivity amongst the three-sulfide solid electrolytes investigated (∼10−2 S cm−1) and showed almost similar charging capacity (when charged to 4.2 V limit) compared to LiNi0.6Mn0.2Co0.2O2 within a conventional liquid carbonate electrolyte (Fig. S1, ESI†).12,28 For the discharge, although each electrode has varying voltage hysteresis, all electrodes show a comparable discharge capacity of around 110 mA h g−1 (compared to 160 mA h g−1 within a liquid electrolyte, Fig. S1, ESI†).
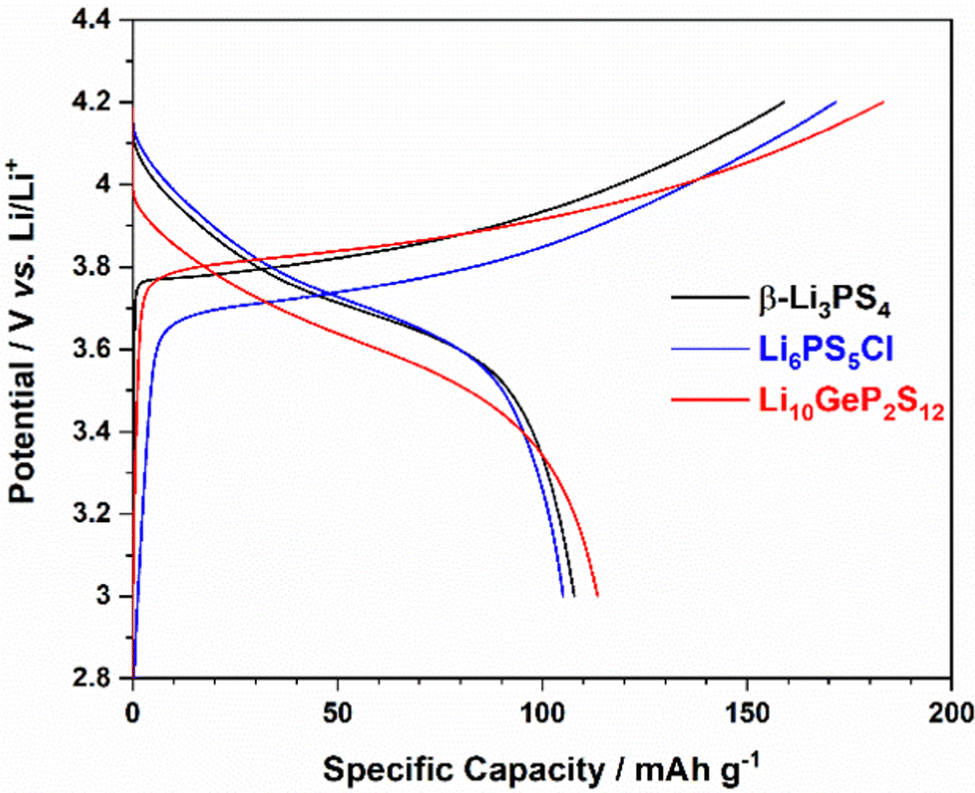 | ||
| Fig. 1 First charge/discharge profile of LiNi0.6Mn0.2Co0.2O2 cycled between 3.0 V and 4.2 V vs. Li/Li+, comprising either β-Li3PS4, Li6PS5Cl or Li10GeP2S12 as the solid electrolyte. | ||
Considering the narrow electrochemical window of all sulfide solid electrolytes,3,4 the interfacial decomposition between solid electrolytes and the active material account for some of the irreversible capacity within the 1st cycle. Since the decomposition of solid electrolytes forms various insulating side products, Raman microscopy was used to identify the chemical nature and type of these products before and after electrochemical cycling.
Raman spectra from all components (LiNi0.6Mn0.2Co0.2O2; conductive additive: vapour grown carbon fibres (VGCF); binder: polyisobutene; solid electrolyte – Fig. S3 and S4, ESI†) of the positive solid-state electrode were collected. Peak assignments are listed within Table 1 and used for the generation of chemical distribution maps. All the samples are recovered and measured under an inert argon atmosphere to avoid air contamination.
Fig. 2 shows typical pristine and cycled spectra for each of the three solid electrolytes containing composite electrodes (refer to Section 1.2 in the ESI† for the composition). Although bands from LiNi0.6Mn0.2Co0.2O2 and sulfide solid electrolytes overlap in the 400–700 cm−1 region, all pristine electrodes show a clearly distinguishable peak at 420 cm−1 ascribed to (PS4)3− from the sulfide solid electrolyte, distinct from the A1g peak of LiNi0.6Mn0.2Co0.2O2 (597 cm−1). The VGCF carbon additives have two distinctive peaks similar to graphitic carbon, the D band and the G− and G+ bands, which are located at ca. 1330 cm−1, 1580 and 1610 cm−1 respectively.17 In addition, uncycled Li6PS5Cl and Li10GeP2S12 electrodes show a negligible amount of Sx (x = 4–8) product at 473 cm−1, which is commonly observed as the chemical decomposition product between sulfide solid electrolytes and metal oxides when they are mixed together.5 After galvanostatic testing, all the electrodes measured showed the presence of new bands, relating to the formation of decomposition products.
 | ||
| Fig. 2 Representative Raman spectra from contour mapping of uncycled and cycled composite electrodes: (a) β-Li3PS4, (b) Li6PS5Cl, and (c) Li10GeP2S12. | ||
In all three cases the main Raman band in the solid electrolyte (vs(PS4)3−) remains detectable, indicating that decomposition is interfacial rather than a bulk degradation process. Firstly, β-Li3PS4 decomposed to the compounds, P2Sx (where x is ca. 5) (380 cm−1) and Sx (473 cm−1).9 For Li6PS5Cl, it initially decomposed to Li3PS4, before forming both P2Sx and Sx. β-Li3PS4 has the (PS4)3− vibration at a similar wavenumber to Li6PS5Cl, which can make the appearance of P2Sx challenging to differentiate from Li6PS5Cl; however, Sx compounds were found from the spectra at 473 cm−1.9 For Li10GeP2S12, Ge–S bonding decomposes into GeS2 and Sx, found in the spectra in Fig. 2c at 348 cm−1 and 473 cm−1.
All spectra show a broad band centred at 1430 cm−1, which cannot be easily assigned by examining the spectra of the individual components (Fig. S3, ESI†). To investigate the origin of this peak, Raman spectra of various slurry mixtures were collected before and after removal of the solvent (Fig. S5 and S6, ESI†). Although slurry samples have strong fluorescence background from organic materials (binder: polyisobutene and solvent: toluene),21 samples containing carbon have a broad 1430 cm−1 band, which is also observed when the dry powders are combined. This suggests that the 1430 cm−1 band arises from reaction of the carbon additive with the solid electrolyte during the electrode preparation. Previously, side reaction between toluene and β-Li3PS4 or Li6PS5Cl has been reported.22,23 To determine whether this is also the case with other sulfide electrolytes, Li10GeP2S12 was mixed within toluene, and GeS2 (348 cm−1) was detected after drying the slurry (Fig. S6, ESI†). Contributions from the binder through vulcanisation reactions or binder degradation were also considered, but these reactions require mild heating (∼150 °C and/or UV irradiation)24,25 and other observations of binder degradation generally occur after electrochemical testing.26,27 Taking this all into account and the absence of the prominent 700 cm−1 band of polyisobutene in any spectra leads to the conclusion that the band appears from reaction between the electrolytes and the carbon additive. Indeed, the band at 1430 cm−1 has been previously reported from the reaction between few-layer graphene and S8.18 This peak is also present in the spectrum of methanethiol (CH3SH) and other linear dithiols19,20 suggesting that the bands arise from CH3 surface functional groups and S linkers, and can be denoted as δ(CH3)–S.
The representative contour mapping image of the β-Li3PS4 based pristine electrode is shown in Fig. 3 and Li6PS5Cl and Li10GeP2S12 contour mappings are available in the ESI† (Fig. S7 and S9). The existence of components from each mapping point was obtained from the spectra and the intensities were normalised. Red colour on the contour scale indicates a relatively intense relevant Raman peak representing the clear presence of a specific component. Black means the absence of the relevant Raman peak above the signal to noise for a specific component at the designated area or spot. From Fig. 3, the location and distribution of LiNi0.6Mn0.2Co0.2O2 and β-Li3PS4 of carbon, based upon their distinctive peaks (listed in Table 1), can be observed. No significant Raman signals of decomposition products resulting from the contact of all materials with the solid electrode were observed.
 | ||
| Fig. 3 Raman contour mapping image of the pristine β-Li3PS4 positive electrode containing active material, carbon additive (VGCF) and electrolyte. Raman contour maps of bands pertaining to (a) LiNi0.6Mn0.2Co0.2O2, (b) β-Li3PS4, (c) the carbon additive and (d) δ(CH3)–S. Red contours represent the presence of intense bands, blue contour trace presence and black, band not detected. | ||
Fig. 4 shows the Raman contour mapping of electrodes after electrochemical cycling. The main bands of β-Li3PS4 electrolyte do not overlap with LiNi0.6Mn0.2Co0.2O2 or the carbon bands, and thereby each component is distinguishable. Moreover, Fig. 4 shows the distribution of decomposition products of β-Li3PS4, P2S5 and Sx from the map. These decomposition products are located between LiNi0.6Mn0.2Co0.2O2 and β-Li3PS4, suggesting that this sulfide SE decomposition is an interfacial reaction. Li10GeP2S12 and Li6PS5Cl have similar decomposition behaviour (Fig. S7–S10, ESI†). In particular, polysulfide (Sx) species were detected on both β-Li3PS4 and Li6PS5Cl solid electrolytes in the vicinity of LiNi0.6Mn0.2Co0.2O2. A similar observation was made with Raman measurements on the LiCoO2 and Li6PS5Cl solid–solid interface,9 highlighting the oxidative stability of the electrolyte breakdowns when in contact with partially de-lithiated positive electrode materials. Though the negative effects of carbon additives on degradation have been frequently reported,29,30 from this initial study no correlation of the degradation products with carbon position was observed. Mapping studies, using a variety of carbons will be probed, as well as in situ mapping investigations to further understand the origin and location of composite electrode degradation. Moreover, to exclude any laser heating effects during electrode mapping, the laser power was minimised to 43 μW. All electrode components were also found to be stable at greater laser intensities (Fig. S11, ESI†).
 | ||
| Fig. 4 Raman contour mapping images of the β-Li3PS4 composite electrode after electrochemical cycling. (a) LiNi0.6Mn0.2Co0.2O2, (b) β-Li3PS4, (c) carbon additive (VGCF), (d) δ(CH3)–S, (e) P2Sx and (f) Sx. | ||
In summary, Raman maps display the distribution of each component within the solid-state electrode at 1–2 micron resolution and show interfacial decomposition between the solid electrolyte and LiNi0.6Mn0.2Co0.2O2. In addition, during the electrode preparation, carbon contamination from sulfide solid electrolyte was found, forming surface δ(CH3)–S. This result highlights the importance of choosing appropriate binder/solvent candidates and lowering the carbon amount present. Both chemical and electrochemical degradation within sulfide solid electrolytes and LiNi0.6Mn0.2Co0.2O2 produces electrically resistive side reaction products, resulting in the lowering of the cell performance, highlighting the need to suppress interfacial decomposition via surface protection.
The authors acknowledge the financial support from the Faraday Institution CATMAT (EP/S003053/1, FIRG016) and SOLBAT (EP/R042047/1, FIRG007) projects. T. A. and S. P. acknowledge the basic funding from the Helmholtz Association.
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. Zhou, N. Minafra, W. G. Zeier and L. F. Nazar, Acc. Chem. Res., 2021, 54, 2717–2728 CrossRef CAS PubMed.
- M. Pasta, D. Armstrong, Z. L. Brown, J. Bu, M. R. Castell, P. Chen, A. Cocks, S. A. Corr, E. J. Cussen, E. Darnbrough, V. Deshpande, C. Doerrer, M. S. Dyer, H. El-Shinawi, N. Fleck, P. Grant, G. L. Gregory, C. Grovenor, L. J. Hardwick, J. T. S. Irvine, H. J. Lee, G. Li, E. Liberti, I. McClelland, C. Monroe, P. D. Nellist, P. R. Shearing, E. Shoko, W. Song, D. S. Jolly, C. I. Thomas, S. J. Turrell, M. Vestli, C. K. Williams, Y. Zhou and P. G. Bruce, J. Phys. Energy, 2020, 2, 032008 CrossRef CAS.
- Y. Nikodimos, C.-J. Huang, B. W. Taklu, W.-N. Su and B. J. Hwang, Energy Environ. Sci., 2022, 15, 991–1033 RSC.
- D. H. S. Tan, E. A. Wu, H. Nguyen, Z. Chen, M. A. T. Marple, J.-M. Doux, X. Wang, H. Yang, A. Banerjee and Y. S. Meng, ACS Energy Lett., 2019, 4, 2418–2427 CrossRef CAS.
- F. Walther, R. Koerver, T. Fuchs, S. Ohno, J. Sann, M. Rohnke, W. G. Zeier and J. Janek, Chem. Mater., 2019, 31, 3745–3755 CrossRef CAS.
- K. Yoon, J.-J. Kim, W. M. Seong, M. H. Lee and K. Kang, Sci. Rep., 2018, 8, 8066 CrossRef PubMed.
- M. Otoyama, Y. Ito, A. Hayashi and M. Tatsumisago, J. Power Sources, 2016, 302, 419–425 CrossRef CAS.
- M. Otoyama, Y. Ito, A. Sakuda, M. Tatsumisago and A. Hayashi, Phys. Chem. Chem. Phys., 2020, 22, 13271–13276 RSC.
- Y. Zhou, C. Doerrer, J. Kasemchainan, P. G. Bruce, M. Pasta and L. J. Hardwick, Batters. Supercaps, 2020, 3, 647–652 CrossRef CAS.
- T. Ates, M. Keller, J. Kulisch, T. Adermann and S. Passerini, Energy Storage Mater., 2019, 17, 204–210 CrossRef.
- K. Chen, S. Shinjo, A. Sakuda, K. Yamamoto, T. Uchiyama, K. Kuratani, T. Takeuchi, Y. Orikasa, A. Hayashi, M. Tatsumisago, Y. Kimura, T. Nakamura, K. Amezawa and Y. Uchimoto, J. Phys. Chem. C, 2019, 123, 3292–3298 CrossRef CAS.
- A. Choi, J. Lim, H.-J. Kim, S. C. Jung, H.-W. Lim, H. Kim, M.-S. Kwon, Y. K. Han, S. M. Oh and K. T. Lee, Adv. Energy Mater., 2018, 8, 1702514 CrossRef.
- Y. Sun, W. Yan, L. An, B. Wu, K. Zhong and R. Yang, Solid State Ion., 2017, 301, 59–63 CrossRef CAS.
- I. Seo and S. W. Martin, Inorg. Chem., 2011, 50, 2143–2150 CrossRef CAS PubMed.
- Q. Pang, X. Liang, A. Shyamsunder and L. F. Nazar, Joule, 2017, 1, 871–886 CrossRef CAS.
- E. Flores, P. Novák and E. J. Berg, Front. Energy Res., 2018, 6 DOI:10.3389/fenrg.2018.00082.
- P. W. Ruch, L. J. Hardwick, M. Hahn, A. Foelske, R. Kötz and A. Wokaun, Carbon, 2009, 47, 38–52 CrossRef CAS.
- C. Bautista-Flores, J. S. Arellano-Peraza, R. Y. Sato-Berrú, E. Camps and D. Mendoza, Chem. Phys. Lett., 2016, 665, 121–126 CrossRef CAS.
- I. W. May and E. L. Pace, Spectrochim. Acta, Part A, 1968, 24, 1605–1615 CrossRef CAS.
- J. Kubackova, I. Izquierdo-Lorenzo, D. Jancura, P. Miskovsky and S. Sánchez-Cortés, Phys. Chem. Chem. Phys., 2014, 16, 11461–11470 RSC.
- L. Cabo-Fernandez, A. R. Neale, F. Braga, I. V. Sazanovich, R. Kostecki and L. J. Hardwick, Phys. Chem. Chem. Phys., 2019, 21, 23833–23842 RSC.
- K. Lee, S. Kim, J. Park, S. H. Park, A. Coskun, D. S. Jung, W. Cho and J. W. Choi, J. Electrochem. Soc., 2017, 164, A2075 CrossRef CAS.
- J. Ruhl, L. M. Riegger, M. Ghidiu and W. G. Zeier, Adv. Energy Sustainability Res., 2021, 2, 2000077 CrossRef CAS.
- K. Lee, J. Lee, S. Choi, K. Char and J. W. Choi, ACS Energy Lett., 2019, 4, 94–101 CrossRef CAS.
- T. Y. Kwon, K. T. Kim, D. Y. Oh, Y. B. Song, S. Jun and Y. S. Jung, Energy Storage Mater., 2022, 49, 219–226 CrossRef.
- R. Black, S. H. Oh, J.-H. Lee, T. Yim, B. Adams and L. F. Nazar, J. Am. Chem. Soc., 2012, 134, 2902–2905 CrossRef CAS PubMed.
- J. Lim, A. Choi, H. Kim, S. W. Doo, Y. Park and K. T. Lee, J. Power Sources, 2019, 426, 162–168 CrossRef CAS.
- A. Choi, J. Lim, H. Kim, S. W. Doo and K. T. Lee, ACS Appl. Energy Mater., 2019, 2, 3427–3434 CrossRef CAS.
- F. Walther, S. Randau, Y. Schneider, J. Sann, M. Rohnke, F. H. Richter, W. G. Zeier and J. Janek, Chem. Mater., 2020, 32(14), 6123–6136 CrossRef CAS.
- E. Quemin, R. Dugas, T. Koç, B. Hennequart, R. Chometon and J.-M. Tarascon, ACS Appl. Mater. Interfaces, 2022, 14(43), 49284–49294 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc01437a |
| ‡ Present address: National Physical Laboratory, Hampton Rd, Teddington, TW11 0LW, UK. |
| This journal is © The Royal Society of Chemistry 2023 |