
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonomeric lithium and sodium silylbenzyl complexes: syntheses, structures, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) O bond olefination†
O bond olefination†
Jordan Barker‡,
Nathan Davison‡
,
Paul G. Waddell and
Erli Lu *
*
Chemistry-School of Natural and Environmental Sciences, Newcastle University, Newcastle upon Tyne NE1 7RU, UK. E-mail: erli.lu@newcastle.ac.uk
First published on 30th May 2023
Abstract
Herein we report the syntheses, structures and reactivity studies of two new monomeric alkali metal silylbenzyl complexes stabilised by a tetradentate amine ligand, tris[2-(dimethylamino)ethyl]amine (Me6Tren). The two complexes, namely [MR′(Me6Tren)] (R′: CH(Ph)(SiMe3)) (2-Li: M = Li; 2-Na: M = Na), exhibit significant different coordination modes according to their metal identity (Li: σ-coordination; Na: π-coordination). Reactivity studies of 2-Li and 2-Na reveal that they are efficient in promoting a widely-used class of organic functional group interconversion: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond olefination of ketones, aldehydes and amides, to produce tri-substituted internal alkenes.
O bond olefination of ketones, aldehydes and amides, to produce tri-substituted internal alkenes.
Organosodium and organolithium complexes play vital roles in synthetic chemistry.1,2 Due to the large ionic radii of Li+ and Na+ and the highly polarised M–C bond (M: Li, Na), these complexes commonly exist as aggregates in solution and solid-state.3–7 It has been long perceived that breaking the aggregates into corresponding monomers could enhance their reactivity.3 From a reaction mechanism perspective,3 the organosodium/organolithium monomers are considered as the key intermediates and the gateway to understanding mechanisms of the reactions mediated by these reagents, as the monomers remove the mingled aggregation and multimetallic effects.
For pursuing the enhanced reactivity (usually referring to Brønsted basicity) and understanding the reaction mechanisms, the coordination chemistry community has invested tremendous efforts in synthesising and characterising the organolithium monomers for decades, from sterically bulky alkyls such as LiCH2SiMe38–11 and tBuLi,12,13 to the least bulky and the archetypical MeLi.14 These research efforts have encompassed the structural features of a number of organolithium monomers, including their essential metal-carbon bond lengths and the bonding characters (e.g., ionicity vs. covalence). Very recently, this interest expanded into organosodium chemistry, represented by an organosodium monomer ([Na(CH2SiMe3)(Me6Tren)], 1-Na) reported by the Hevia15 and our16 groups, simultaneously and independently, in 2023, which exhibited not only enhanced but also unique reactivity.
In comparison with the relatively well-documented synthesis and structural studies, the reactivity of organolithium/organosodium monomers is less studied, and almost all the reported reactivity studies (bar for 1-Na16) focused on Brønsted basicity (deprotonation) (e.g., ref. 9–11) and the follow-up nucleophilic substitution (such as aroylation15). Recently, we reported a rare non-deprotonation reactivity pattern, where [Na(CH2SiMe3)(Me6Tren)] (1-Na) conducted CO bond methylenation.16 This case highlighted the hitherto largely unexplored versatile reactivity scope of the organolithium and organosodium monomers.
To unlock the exciting chemical space, it is essential to expand the alkyl group scope of the organolithium/organosodium monomers. Herein, we introduce silylbenzyl, namely [CH(SiMe3)Ph]− (R′), into organolithum/organosodium monomer chemistry (despite that phenyl-substituted silyl benzyls are known in Group-1/2 chemistry17–23), reporting syntheses of the monomers [MR′(Me6Tren)] (M = Li: 2-Li; M = Na; 2-Na), which exhibit diversified coordination modes between the metal cation and the R′ alkyl. Moreover, 2-Li/Na can convert the CO bond in ketones, aldehydes and amide into CC(H)(Ph) bond, i.e., conducting the CO bond olefination. These results are communicated herein.
The target monomers, 2-Li and 2-Na, can be synthesised by treating their corresponding [M(CH2SiMe3)(Me6Tren)] monomer precursors (1-M; M = Li,11 Na16) (formed in-situ by treating [MCH2SiMe3]n with one equivalent of Me6Tren; See ESI† for more details) with PhCH2SiMe3, respectively (Scheme 1). Single crystals suitable for X-ray diffraction studies were grown from their hexane/benzene mixed solutions under −35 °C, and their SCXRD molecular structures are displayed in Fig. 1.
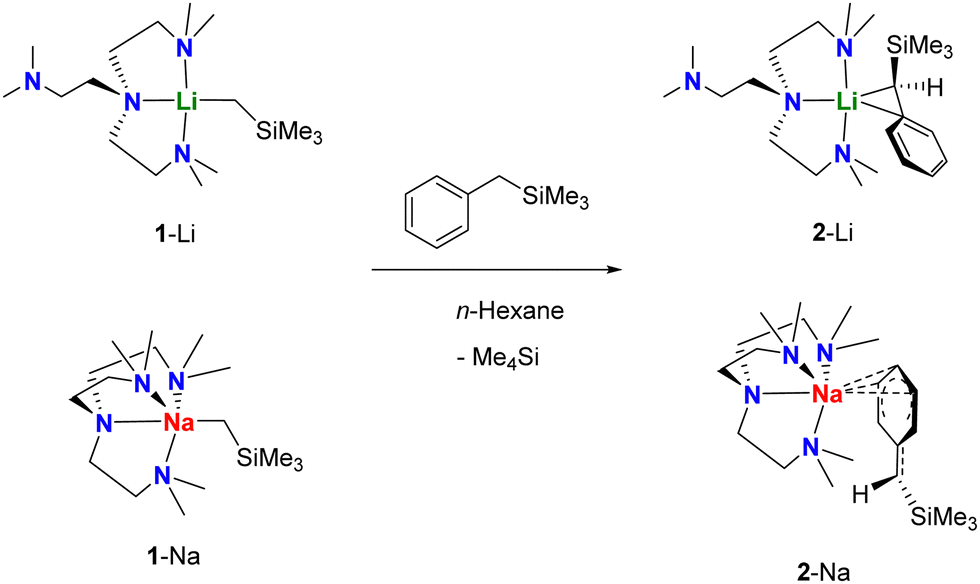 | ||
| Scheme 1 Syntheses of lithium and sodium silylbenzyl monomers 2-Li and 2-Na. | ||
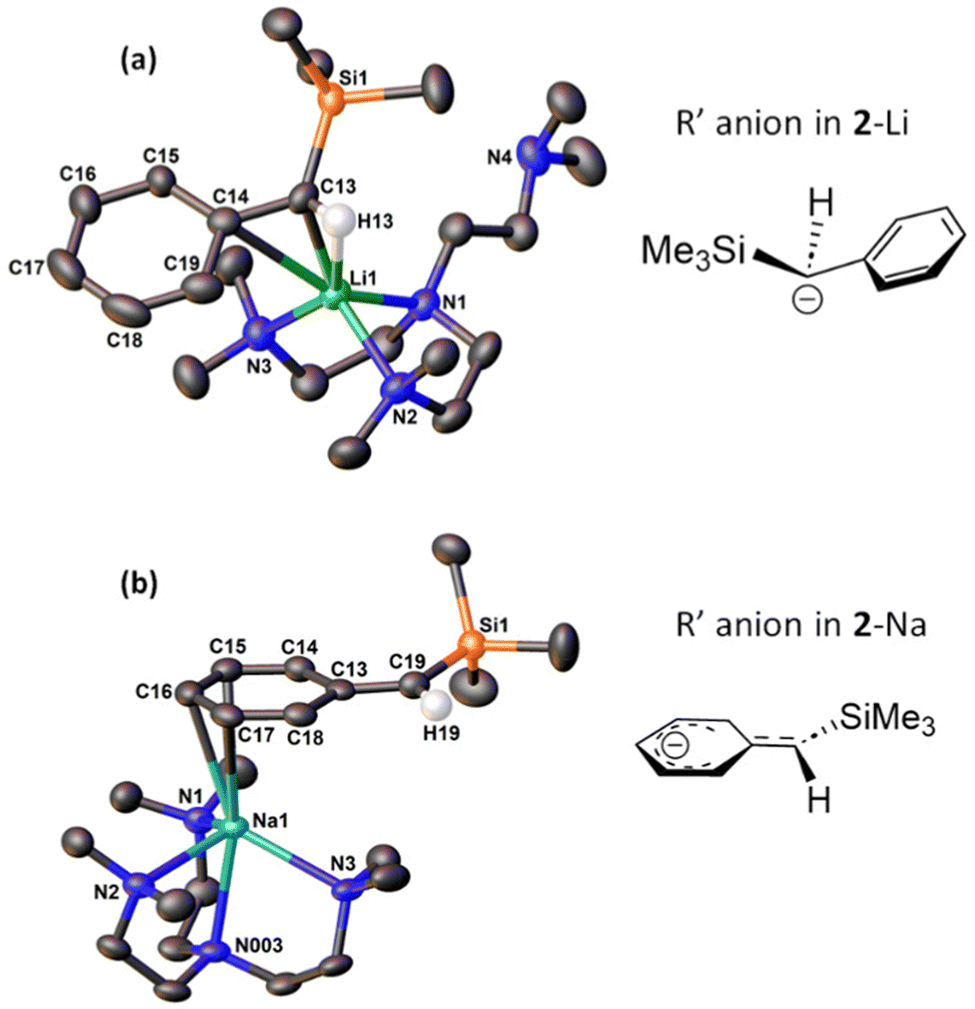 | ||
| Fig. 1 Single-crystal X-ray diffraction (SCXRD) molecular structures of 2-Li (a) and 2-Na (b) measured at 150 K. Protons (except the benzylic protons) and solvent molecules in the lattice are omitted for clarity. Schematic representations of their R′ anion are displayed next to the corresponding SCXRD structure. Key bond distances (Å): 2-Li: Li1–C13 2.219(3); Li1–C14 2.759(3); Li1–N1 2.182(3); Li1–N2 2.197(3); Li1–N3 2.090(3); Li1–H13 2.322(17); C13–C14 1.437(2); C13–H13 0.940(17). 2-Na: Na1–C15 3.0017(15); Na1–C16 2.8179(15); Na1–C17 2.9066(15); Na1–N1 2.5949(12); Na1–N2 2.5834(12); Na1–N3 2.4849(12); Na1–N003 2.5466(12); C13–C14 1.429(2); C14–C15 1.372(2); C15–C16 1.399(2); C16–C17 1.392(2); C17–C18 1.383(2); C18–C13 1.4469(19); C13–C19 1.4038(19). | ||
The salient structural feature of 2-Li and 2-Na is their different coordination modes of the silylbenzyl (R′) group. In 2-Li, the R′ group bonds to Li+ mostly through the benzylic carbon atom (C13) via a η1-C coordination, with a Li1–C13 bond length of 2.219(3) Å. This is significantly shorter than the Li–Cbenzylic bond length (2.352(3) Å) in a benzyl lithium monomer [Li(η1-CH2Ph)(Me6Tren)] reported by Robertson, Mulvey and co-workers.24 We also observed a relatively weak interaction between the ipso-carbon (C14) and Li+ (Li1–C14 2.759(3) Å). The bond length between the benzylic-carbon (C13) and ipso-carbon (C14) is 1.437(2) Å, indicating a typical C–C single bond. Correspondingly, the geometry of the benzylic carbon C13 is best described as a distorted tetrahedron, with a sum of angles (Σ {∢ Si1–C13–C14, C14–C13–H13, H13–C13–Si1}) of 349.5°.
On the other hand, in 2-Na, the R′ group bonds to Na+ through three carbon atoms (C15, C16, C17) in the phenyl ring, rendering the coordination mode to be η3-C3. The Na–C bond lengths are relatively short (2.8 to 3.0 Å): they are at least 0.2 Å shorter than the Na–Cipso distance (3.183(1) Å) in Robertson/Mulvey's benzyl sodium monomer [Na(η2-CH2Ph)(Me6Tren)],24 where the Na–Cipso interaction was reported to be pronounced. Hence, we conclude here that, in 2-Na, the R′ silylbenzyl group coordinates to Na+ through three strong Na–CPh bonds. It is intriguing to investigate more structural details of the R′ group in 2-Na. The C–C bond lengths in the phenyl ring exhibit a relatively wide distribution, and divide into two groups: one group is close to the expected value for delocalised arenes (∼1.39 Å, C14–C15; C15–C16; C16–C17; C17–C18), while the other group features significantly longer carbon-carbon bonds (1.43-1.44 Å. C13–C14; C18–C13). Meanwhile, the Cipso–Cbenzylic bond length (C13–C19 1.4038(19) Å) is in the CC double bond regime, and the Cbenzylic (C19) features an alkene-type planar geometry (Σ∡ = 360°).
It is well known that different alkali metal cations prefer different σ- or π-coordination modes. The σ- or π-affinities are best demonstrated in benzyl or substituted benzyl complexes, since the anions offer both σ- (the benzylic carbon) and π-(phenyl) sites. In general, the light alkali metal cations Li+ and Na+ are more likely to adopt the σ-coordination mode, i.e., the metal cation coordinates to the benzylic carbon, possibly accompanied by weak interaction(s) with the ipso-carbon. On the other hand, heavier alkali metal cations (K+, Rb+, Cs+) are more likely to adopt the π-coordination mode, i.e., the metal cation coordinates to the phenyl ring. The trend can be clearly seen in the 2011 systematic study of alkali metal benzyl complexes24 by Mulvey, Robertson and co-workers, and a 2022 follow-up from the same groups, expanding the anion scope into relevant ditopic arylmethyl anions such as diphenyl methyl and fluorenyl.25 In comparison with the generally similar Li+/Na+ coordination mode in literature (especially compared with the Robertson/Mulvey's [Li/Na(CH2Ph)(Me6Tren)] monomers24), in 2-Li and 2-Na the Li+ and Na+ exhibit entirely different coordination modes, which we attribute to the increased steric congestion of the silyl benzyl R′ compared with benzyl. It should be noted that the σ- or π-preference trend of alkali metal cations is not rigid and there are exceptions where Li+/Na+ adopt π-coordination mode (e.g., with fluorenyl25) and K+/Rb+/Cs+ adopt σ-coordination mode (e.g., with -NMe2 substituted silyl benzyl18), usually caused by overriding electronic (e.g., for the fluorenyl) or steric (e.g., for the -NMe2 substituted silyl benzyl) factors.
From a reactivity perspective, converting CO bond into CC bond, i.e., carbonyl olefination, is an essential class of organic functional group interconversions.26 The popular olefination methods include Wittig,27 Tebbe,28 Julia29/Julia-Kocieński,30 Peterson31 and so on. All these methods require hazardous/expensive reagents (e.g., phosphorus ylide for Wittig, sulfones for Julia, titanium for Tebbe, cerium for modified Peterson) and/or conditions (e.g., strong acids/bases for Peterson/modified Peterson).
We recently reported that the –SiMe3 group in 1-Na can serve as the leaving group, enabling the NaCH2SiMe3 to act as a [CH2] feedstock for CO bond methylenation.16 We are intrigued to expand this methodology to internal alkenes, utilising 2-Li/Na as [CHPh] feedstock, realising CO bond olefination to produce internal alkenes. Moreover, most traditional olefination methods perform less well for sterically congested substrates (especially for Wittig reagents due to their four-membered ring transition state). Since we observed a preference of the steric-bulky substrates in our previous methylenation work,16 expanding the methodology into constructing sterically congested internal alkenes would be attractive.
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Reactions between 2-Li/Na and nine ketones/aldehydes/amides (3a–i) were studied, and the results are summarised in Table 1. In eight out of the nine cases, 2-Li/Na promoted smooth olefinations under mild conditions. Unlike our previous methylenation report,16 we do not observe different reaction patterns between 2-Li and 2-Na, despite their structural distinction. We attribute the same reaction pattern of 2-Li and 2-Na to their increased steric congestion compared with the -CH2SiMe3 (R) counterparts 1-Li/Na.16 In the previous report,16 DFT reaction pathway calculations suggested that the key step, i.e., intramolecular –SiMe3 elimination, prefers sterically bulky environment. The bulky CH(Ph)SiMe3 (R′) group would likely facilitate the key −SiMe3 elimination step to a level that both Li and Na's kinetic barriers are low enough for the olefination to proceed.
:1 Reactions between 2-Li/Na and nine ketones/aldehydes/amides (3a–i) were studied, and the results are summarised in Table 1. In eight out of the nine cases, 2-Li/Na promoted smooth olefinations under mild conditions. Unlike our previous methylenation report,16 we do not observe different reaction patterns between 2-Li and 2-Na, despite their structural distinction. We attribute the same reaction pattern of 2-Li and 2-Na to their increased steric congestion compared with the -CH2SiMe3 (R) counterparts 1-Li/Na.16 In the previous report,16 DFT reaction pathway calculations suggested that the key step, i.e., intramolecular –SiMe3 elimination, prefers sterically bulky environment. The bulky CH(Ph)SiMe3 (R′) group would likely facilitate the key −SiMe3 elimination step to a level that both Li and Na's kinetic barriers are low enough for the olefination to proceed.
O bond olefinations
|
|
||||
|---|---|---|---|---|
| Olefination product(s) | Conditions | 2-Li | 2-Na | |
| Conversion (%); E/Z ratio | Conversion (%); E/Z ratio | |||
 |
 |
60 °C 2 hours | > 95% | > 95% |
 |
 |
R. T. 30 min | > 95% 2:3 |
> 95% 1:1 |
 |
 |
RT 30 min | > 95% | > 95% |
 |
 |
RT 30 min | > 95% 2:3 |
> 95% 1:1 |
 |
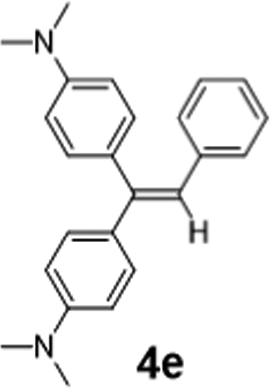 |
RT 20 hours | > 95% | > 95% |
 |
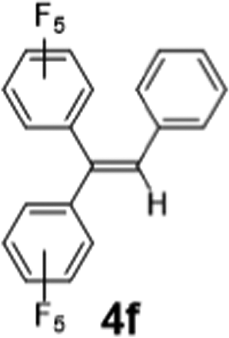 |
RT 30 min | Intractable mixture | Intractable mixture |
 |
 |
RT 30 min | > 95% 3:2 |
> 95% 1:1 |
 |
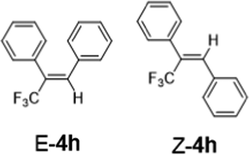 |
60 °C 2 hours | > 95% 1:2 |
> 95% 1:3 |
 |
 |
RT 30 min | > 95% 2:3 |
> 95% 2:3 |
As a preliminary Communication, we do not aim to cover a comprehensive substrate scope here. Nevertheless, 2-Li/Na perform well with a range of ketones, aldehydes and an amide (Table 1). tri-Substituted internal alkenes, such as 4a–i, are challenging targets for traditional olefination methods such as Wittig27 and Julia–Kocieński29,30 reagents, which prefer steric less bulky substrates. In contrast, 2-Li/Na promote olefinations of these steric bulky substrates with high conversions under mild conditions, providing a valuable route to synthesise the tri-substituted internal alkenes. Interestingly, mild E/Z selectivity was observed for 4b, 4g, 4h and 4i, which implies an interesting possibility of using chiral ligands to manipulate the selectivity.
In conclusion, we reported the syntheses and characterisations of lithium and sodium silylbenzyl monomers 2-Li and 2-Na, revealing their pronounced structural differences regarding the coordination modes of the silylbenzyl (R′) group. While Li+ coordinates to R′ via an unsurprising σ-interaction, Na+ behaves unexpectedly and adopts an entire π-coordination. Reactivity studies of 2-Li/Na towards organic carbonyl substrates demonstrate their capabilities to deliver olefinations, converting the CO bond into tri-substituted internal alkenes. The olefination reactivity complements our previous report on NaCH2SiMe3-mediated ketone/aldehyde methylenation and provides a less hazardous alternative for the traditional olefination reagents such as Wittig, Tebbe, Julia/Julia-Kocieński, Peterson and so on.
This work demonstrates the immense potential of Group-1 metal organometallic chemistry beyond the well-perceived deprotonation/nucleophilic/metal-exchange reactivity scope. Further work is underway in our group, focusing on exploiting the olefination reactivity and our recently reported ligand catalysis strategy,16 aiming at realising organo-alkali metal-mediated E/Z selective olefination catalysed by feasible chiral amine ligands.
The authors thank the Newcastle University Chemistry Technical Support Team (Dr Laura McCorkindale, Dr Amy Roberts and Mr Niall Straughan) for supporting our research. E. L. thanks the Newcastle University Academic Track (NUAcT) Fellowship Scheme, EPSRC North East Centre of Energy Materials (NECEM) and the Royal Society of Chemistry Research Enablement Grants (E20-5153) for financial support. N. D. thanks Newcastle University for a NUAcT PhD studentship and the Royal Society of Chemistry Research Enablement Grants (E22-3348740748). J. B. thanks Newcastle University for the financial support of his final-year MChem research project in the E. L. group.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- Z. Rappoport and I. Mare, The chemistry of organolithium compounds, John Wiley & Sons, Ltd, Chichester, West Sussex, England, 2004 Search PubMed.
- P. B. De, S. Asako and L. Ilies, Synthesis, 2021, 3180–3192, DOI:10.1055/a-1478-7061.
- H. J. Reich, Chem. Rev., 2013, 113, 7130–7178, DOI:10.1021/cr400187u.
- V. H. Gessner, C. Däschlein and C. Strohmann, Chem. – Eur. J., 2009, 15, 3320–3334, DOI:10.1002/chem.200900041.
- A. Harrison-Marchand and F. Mongin, Chem. Rev., 2013, 113, 7470–7562, DOI:10.1021/cr300295w.
- F. Mongin and A. Harrison-Marchand, Chem. Rev., 2013, 113, 7563–7727, DOI:10.1021/cr3002966.
- R. A. Gossage, J. T. B. H. Jatrzebski and G. van Koten, Angew. Chem., Int. Ed., 2005, 44, 1448–1454, DOI:10.1002/anie.200462103.
- M. F. Lappert, L. M. Engelhardt, C. L. Raston and A. H. White, J. Chem. Soc., Chem. Commun., 1982, 1323–1324, 10.1039/C39820001323.
- T. Tatic, H. Ott and D. Stalke, Eur. J. Inorg. Chem., 2008, 3765–3768, DOI:10.1002/ejic.200800610.
- L. Knauer, J. Wattenberg, U. Kroesen and C. Strohmann, Dalton Trans., 2019, 48, 11285–11291, 10.1039/C9DT02182E.
- N. Davison, P. G. Waddell, C. Dixon, C. Wills, T. J. Penfold and E. Lu, Dalton Trans., 2022, 51, 10707–10713, 10.1039/D1DT03532K.
- C. Strohmann, T. Seibel and K. Strohfeldt, Angew. Chem., Int. Ed., 2003, 42, 4531–4533, DOI:10.1002/anie.200351308.
- C. Strohmann and V. H. Gessner, Angew. Chem., Int. Ed., 2007, 46, 8281–8283, DOI:10.1002/anie.200702116.
- N. Davison, E. Falbo, P. G. Waddell, T. J. Penfold and E. Lu, Chem. Commun., 2021, 57, 6205–6208, 10.1039/D1CC01420J.
- D. E. Anderson, A. Tortajada and E. Hevia, Angew. Chem., Int. Ed., 2023, 62, e202218498, DOI:10.1002/anie.202218498.
- N. Davison, C. L. McMullin, L. Zhang, S.-X. Hu, P. G. Waddell, C. Wills, C. Dixon and E. Lu, J. Am. Chem. Soc., 2023, 145, 6562–6576, DOI:10.1021/jacs.3c01033.
- L. T. Byrne, L. M. Engelhardt, G. E. Jacobsen, W.-P. Leung, R. I. Papasergio, C. L. Raston, B. W. Skelton, P. Twiss and A. H. White, J. Chem. Soc., Dalton Trans., 1989, 105–113, 10.1039/DT9890000105.
- F. Feil and S. Harder, Organometallics, 2001, 20, 4616–4622, DOI:10.1021/om010444j.
- C. Strohmann, D. H. M. Buchold, T. Seibel, K. Wild and D. Schildbach, Eur. J. Inorg. Chem., 2003, 3453–3463, DOI:10.1002/ejic.200300323.
- B. Jia, X. Wei, H. Tong, M. Zhou and D. Liu, Inorg. Chim. Acta, 2012, 388, 127–134, DOI:10.1016/j.ica.2012.02.022.
- H. Schumann, D. M. M. Freckmann and S. Dechert, Z. Anorg. Allg. Chem., 2008, 634, 1334–1338, DOI:10.1002/zaac.200800067.
- H. Ott, C. Daschlein, D. Leusser, D. Schildbach, T. Seibel, D. Stalke and C. Strohmann, J. Am. Chem. Soc., 2008, 130, 11901–11911, DOI:10.1021/ja711104q.
- S. G. Koller, U. Kroesen and C. Strohmann, Chem. – Eur. J., 2015, 21, 641–647, DOI:10.1002/chem.201405152.
- M. G. Davidson, D. Garcia-Vivo, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Chem. – Eur. J., 2011, 17, 3364–3369, DOI:10.1002/chem.201003493.
- A. Rae, K. M. Byrne, S. A. Brown, A. R. Kennedy, T. Kramer, R. E. Mulvey and S. D. Robertson, Chem. – Eur. J., 2022, 28, e202104260, DOI:10.1002/chem.202104260.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley-Blackwell, 6th Edn, 2007 Search PubMed.
- G. Wittig and U. Schöllkopf, Chem. Ber., 1954, 87, 1318–1330, DOI:10.1002/cber.19540870919.
- F. N. Tebbe, G. W. Parshall and G. S. Reddy, J. Am. Chem. Soc., 1978, 100, 3611–3613, DOI:10.1021/ja00479a061.
- M. Julia and J.-M. Paris, Tetrahedron Lett., 1973, 14, 4833–4836, DOI:10.1016/S0040-4039(01)87348-2.
- P. R. Blakemore, W. J. Cole, P. J. Kocieński and A. A. Morley, Synlett, 1998, 26–28, DOI:10.1055/s-1998-1570.
- L. F. van Staden, D. Gravestock and D. J. Ager, Chem. Soc. Rev., 2002, 31, 195–200, 10.1039/A908402I.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2250196 and 2250197. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc01376f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |