
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterobimetallic 21,23-dimetallaporphyrin: activation of metal–metal interactions within the porphyrinoid macrocycle†
Grzegorz
Vetter
 ,
Agata
Białońska
,
Aneta
Jezierska
,
Jarosław J.
Panek
and
Ewa
Pacholska-Dudziak
*
,
Agata
Białońska
,
Aneta
Jezierska
,
Jarosław J.
Panek
and
Ewa
Pacholska-Dudziak
*
Department of Chemistry, University of Wrocław, Wroclaw, Poland. E-mail: ewa.dudziak@uwr.edu.pl
First published on 15th May 2023
Abstract
Two core-modified porphyrins containing metal atoms, namely platinum(II) or platinum(IV) and rhodium(III), in place of two NH units, have been obtained by a post-synthetic modification of the 21,23-ditelluraporphyrin. The products of the tellurium-to-metal exchange, 21-platina-23-rhodaporphyrins, incorporate rhodacyclopentadiene and platinacyclopentadiene units with the metal atoms facing each other. The two molecules exhibit different degrees of metal–metal interaction depending on the oxidation state of platinum, with the NBO bond order being 0.04 for platinum(IV) and 0.15 for platinum(II). Consistently, the Quantum Theory of Atoms in Molecules analysis revealed the presence of the bond determinant, the bond critical point, in the platinum(II) species, in contrast to the platinum(IV) congener. The two porphyrinoids are interconvertible in redox reactions. They both exhibit fluxional behaviour in solution, studied by 1H NMR, involving alteration in the metal ion coordination sphere accompanied by the macrocyclic skeleton conformation change.
Metal–metal interaction is an important and broad concept, attracting unabated attention.1–3 Metal–metal bonds break new records, reaching orders as high as five4 and even six,5 while on the other end of the scale, the weak, metallophilic interactions6 and bimetallic frustrated pairs7 turn out to be no less fascinating. The metal–metal-bonded compounds find applications as structural subunits of metal–organic frameworks, molecular-scale conductors, photosensitisers, and catalysts. In organometallic catalysis, the possible cooperativity and synergy of the two metals placed in proximity give advantages by means of possible multi-site activation, bimetallic preorganisation, fine-tuning of the metal properties, or acting as an electron reservoir.8 The porphyrin macrocycle is not typically associated with bimetallic complexes. However, the metal–metal interactions of various order, dependent on the central atoms’ electronic configuration and the macrocycles’ spatial relation, are present in this area of research in dimeric structures.9–11 Apart from a rich collection of coordination compounds with metal–metal bonds extending over a porphyrin ring,12 a single porphyrin macrocycle has rarely been used as a platform for a metal–metal bond investigation. It was, however, possible for Co(II)⋯Ru(0) interactions in a unique paramagnetic σ/π-hybrid complex of azuliporphyrin.13 Dinaphthoporphycene with an elongated N4 core was capable of binding two palladium(II) ions in an unprecedented cis-mode, exhibiting significant metal–metal bonding interaction.14
The potential of porphyrinoid chemistry to construct organometallic binuclear species with two metal ions driven to close proximity has been discovered recently. The synthesis of organometallic aromatic 21,23-dirhodaporphyrin 1 containing two rhodium(III) ions has been reported.15 The two metal atoms were incorporated into the porphyrin skeleton in place of two NH groups. The bridged Rh2Cl2 unit situated approximately within the macrocyclic plane contains two 18-electron rhodium(III) centres surrounded by octahedral environments. The 21,23-dimetallaporphyrin 1 is formally derived from a porphyrin-annulene hybrid, 21,23-divacataporphyrin, 2,16,17 however, the synthetic route started from 21,23-ditelluraporphyrin 318,19 and involved two heteroatom, tellurium to rhodium, substitution steps (Chart 1).
 | ||
| Chart 1 Dimetallaporphyrin 1, its formal ligand 2, and its synthetic precursor 3. | ||
Herein, we present studies on heterobimetallic compounds, 5 and 5-Cl2, containing rhodium(III) and platinum(II) or platinum(IV), constructed of two metallacyclopentadiene units and two pyrroles, coined into an organometallic aromatic system. The synthesis avoided unlikely four-fold activation of C–H bonds of the formal ligand, 2, and followed the tellurium-to-metal substitution protocol, employed in our former studies.15,20–22 The synthetic path, starting with tetrakis(4-methoxyphenyl)-21,23-ditelluraporphyrin 3,16 takes advantage of a significant difference in the reactivity of a tellurophene unit towards platinum(II) and rhodium(I) salts, which determines the order of steps. Thus, a single tellurium-to-metal replacement proceeded with platinum(II), yielding the known 21-platina-23-telluraporphyrin 4,21 which served as a starting material for the second substitution with the more reactive rhodium(I), as shown in Scheme 1. Reordering of the reaction steps did not provide the desired product, since a tellurium-to-platinum replacement has never occurred for 21-rhoda-23-telluraporphyrin or any other 21-metalla-23-telluraporphyrin.
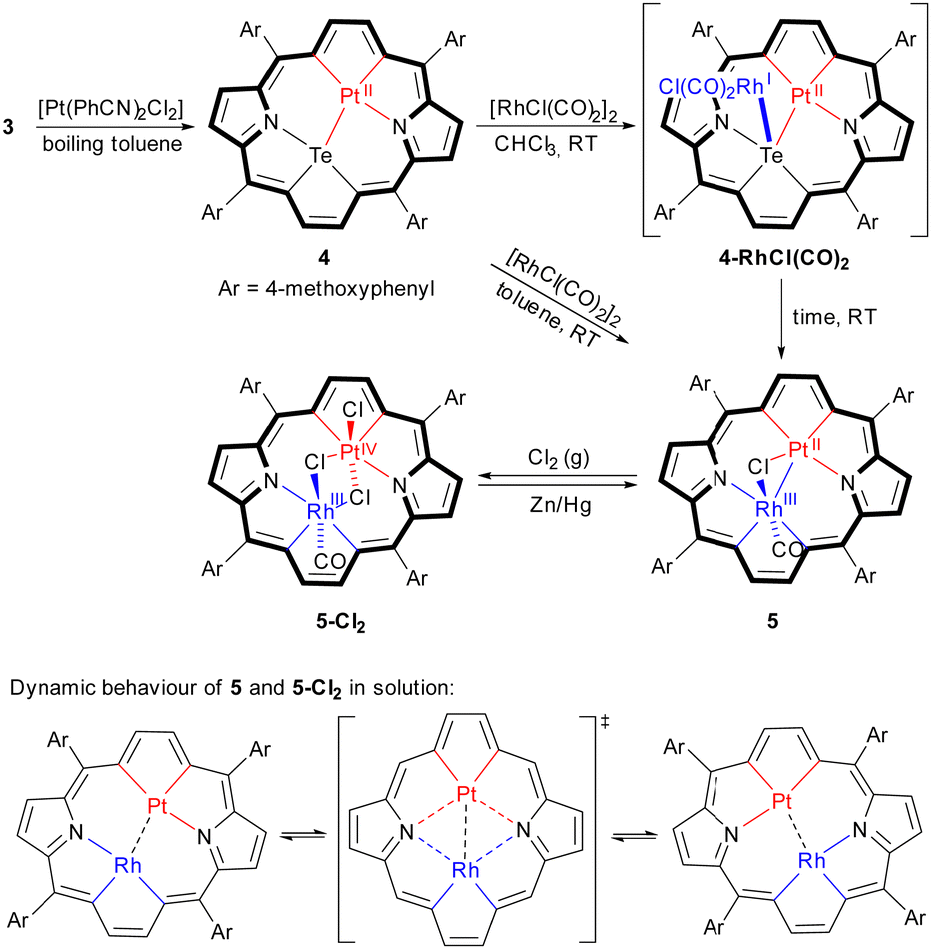 | ||
| Scheme 1 Synthesis of 21-platina-23-rhodaporphyrins and their dynamic behaviour in solution; in the bottom scheme, axial ligands are omitted for clarity. | ||
Use of toluene as the medium for the reaction of 4 with [RhCl(CO)2]2 promoted the direct formation of the desired 21-platina-23-rhodaporphyrin, 5, while application of chloroform allowed for spectroscopic (1H NMR and UV-Vis) detection of a moderately stable coordination compound, which subsequently transforms into 5 on standing in solution within days. The formula 4-RhICl(CO)2 postulated for this intermediate is based on the detection of cationic species 4-Rh(CO)2+ in mass spectrometry and supported by DFT calculations (ESI†). Adducts with [Rh(CO)2Cl] fragments attached to one donor atom are known in porphyrinoid chemistry.23,24 Generally, it can be considered as a rule, that the tellurium-to-metal substitution in metallaporphyrin synthesis is preceded by the formation of a side-on coordination compound.15,20–22 The heterobimetallic 5, reacts with chlorine through an oxidative addition at the platinum(II) centre yielding quantitatively the platinum(IV) analogue, 5-Cl2. The reaction can be readily reversed with zinc amalgam.
The two heterobimetallic 21,23-dimetallaporphyrins were characterized in the solid state by X-ray crystallography and in solution by means of NMR and UV-Vis spectroscopy (Fig. 1). The platinum(II) species, 5, has also been characterised by electrochemistry (Fig. 2). The molecular structures of 5 and 5-Cl2 (Fig. 3), apart from a moderate out of plane distortion, exhibit the characteristic rhomboidal shape of macrocyclic skeletons, imposed by the central metal ions’ coordination preferences and their relatively large size. Such a macrocyclic in-plane deformation has been previously observed for 21-metalla-23-telluraporphyrins20,21 and for the 21,23-dirhodaporphyrin 1.15 Significantly, the metal atoms in 21-platina-23-rhodaporphyrins are accommodated practically within the macrocyclic plane. The deformation of the skeletons of 5 and 5-Cl2 did not significantly disturb the delocalization within the 18 π-electron conjugation path of the porphyrinoid C20N2 skeleton, shown with the bold line in Scheme 1.
 | ||
| Fig. 1 UV-Vis absorption spectra (CH2Cl2 solutions) of 5 (solid black line) and 5-Cl2 (dashed red line). | ||
 | ||
| Fig. 2 Cyclic voltammograms (top) and differential pulse voltammograms (bottom) for 5. The arrows indicate the directions of the electrode potential advancements; the electrode potentials in volts are given in green. The red asterisk indicates the reduction of the impurity generated in the irreversible second oxidation at 0.82 V (CV). The electrochemical HOMO–LUMO gap (HLG = EOx1 − ERed1) equals 1.58 V. | ||
 | ||
| Fig. 3 Top: X-Ray molecular structures of 5 and 5-Cl2; thermal ellipsoids represent 50% probability. Bottom: QTAIM analysis results for 1, 5 and 5-Cl2, green – bond critical points (BCP), red – ring critical points (RCP); bond paths – black lines; low electron density at BCP is signaled by a dashed line. | ||
The electronic spectra, with intense Soret-like bands at 500 and 502 nm for 5 and 5-Cl2, respectively (Fig. 1), support their macrocyclic aromaticity, which is also manifested in the 1H NMR spectra as prominent chemical shifts (Fig. S3 and S7, ESI†). Thus, β-pyrrole hydrogen signals appear at 8.65–8.32 ppm in 5 and 8.59 ppm in 5-Cl2 at 300 K while β-metallacyclopentadiene protons exhibit significantly larger δ values (5: 10.07–9.45 ppm; 5-Cl2: 10.08–9.61 ppm; 300 K), similar to those in the known metallaporphyrins.15,20,21 These resonances are readily distinguished on the basis of heteronuclear couplings with NMR-active nuclei, 103Rh and 195Pt. The rhodacyclopentadiene signals were split with small coupling constants by rhodium-103 (3JRhH = 1.5 Hz for 5; 1.2 Hz for 5-Cl2), while the interaction with platinum-195 (37% abundance; 3JPtH = 51 Hz for 5-Cl2) has been detected as broad satellites present for platinum(IV) species only, while for platinum(II) the severe broadening due to the chemical shift anisotropy relaxation impeded the detection of the lines.25 Lack of molecular symmetry elements, revealed in the solid state structure, renders all the porphyrin perimeter protons inequivalent, thus, provided a similar structure is maintained in solution, eight β-hydrogen signals are expected for both 21,23-dimetallaporphyrins. While the 1H NMR spectrum of 5 in standard conditions (300 K) was in accordance with such a pattern, for 5-Cl2, the temperature had to be lowered to 180 K to observe the eight resonances. The room temperature spectrum of 5-Cl2 corresponds with a two-fold molecular symmetry due to dynamic averaging and it includes one pyrrole AB pattern, one 103Rh-split doublet and one singlet flanked by broad 195Pt satellites. The dynamic behaviour, depicted in Scheme 1, responsible for these spectral features has been studied by means of variable-temperature 1H NMR studies and supported by DFT calculations (Fig. S4, S7 and S15, ESI†). In fact, both studied 21,23-dimetallaporphyrins, 5 and 5-Cl2, exhibit fluxionality in solution, although they differ in the kinetics. The molecular movement involves changes in the coordination sphere of both metal centres, switching between two nitrogen atoms, N22 and N24, while the other bonds around central ions are maintained. These changes concord with the conformation changes of the flexible macrocyclic frame. Such behaviour is analogous to that of 21,23-dirhodaporphyrin 1, and several 21-metalla-23-telluraporphyrins.15,20,21 The platinum(II)-containing 5 exhibits slower motion, which is reflected by higher activation energy (ΔG‡ = 17.4(5) kcal mol−1, as estimated from the coalescence temperature) compared to 5-Cl2 (ΔG‡ = 9.4(2) kcal mol−1). The significantly faster exchange for platinum(IV) as compared with platinum(II) species 5 is in accordance with the trends observed for fluxional platinum complexes.21,26 The analogous dynamic process observed for 21,23-dirhodaporphyrin 1, was too fast to allow the registration of a 1H NMR spectrum in the slow exchange limit (DFT calculated Ea = 6.2 kcal mol−1).
A closer examination of the molecular structures shows significant differences between 5 and 5-Cl2 in the bonding within the central bimetallic unit. While in 5-Cl2 both central ions, rhodium(III) and platinum(IV), exhibit expected octahedral environments, provided by C3NCl2 and C2NCl3 donors, respectively, in 5, both ions have atypical surroundings. For platinum(II), the anticipated square planar environment, although deformed, is fulfilled by C2NCl donors, with a relatively long Pt–Cl distance of 2.486(4) Å and angles far from 90° (Fig. S12, ESI†). The rhodium(III) centre, however, is surrounded by only five nonmetals, C3NCl, while the sixth post is occupied by the neighbouring metal, platinum(II). The severely deformed octahedron involves the metal atom within the coordination sphere.
In order to examine the possible metal–metal interactions, we performed DFT calculations (ωB97-D3/def2-TZVP) followed by the Quantum Theory of Atoms in Molecules (QTAIM), Natural Bond Orbital (NBO), and Electron Localization Function (ELF) analyses. For the sake of comparison, we also included 21,23-dirhodaporphyrin 1 in the theoretical studies. We examined this molecule experimentally in our previous work,15 where we claimed an absence of a rhodium-rhodium bond on the basis of lower level preliminary calculations. The analysis performed in the present work for 1, 5 and 5-Cl2 (see the ESI† for discussion and computational details) suggests the presence of a weak Pt(II)–Rh(III) bond in 5, but no evidence of metal–metal bonding is found for 1 or 5-Cl2. In particular, the QTAIM reveals metal–metal Bond Critical Points (BCPs) for 1 and 5, but not for 5-Cl2 (Fig. 3). However, the values of electron density and ellipticity at the BCPs (1: ρ 0.0264 a.u., ellipticity: 1.665 vs.5: ρ: 0.0463 a.u., ellipticity: 0.190) indicate that this BCP is not a signature of a metal–metal interaction in 1 but rather a consequence of the spatial proximity of the metal centres, resulting in the formation of a BCP solely to satisfy the topological Poincaré–Hopf theorem. The electron density at the metal–metal bond path, BCP in 5 (0.0463 a.u.) shows the same order as the values for non-metal–metal bonds around rhodium and platinum atoms (from 0.0623 for Pt–Cl to 0.1077 a.u. for Pt–N, Table S3, ESI†). QTAIM results correspond with NBO analysis outcomes: the Wiberg bond index calculations for the metal–metal bond, giving 0.153 for 5, the value, while not close to 1, as for an ideal single bond, still markedly higher than for the other considered molecules (1: 0.0405, 5-Cl2: 0.032). It is also very close to the value of 0.18 found for the Pd–Pd bond in a cis-bipalladium complex of dinaphthoporphycene.14 Moreover, ELF calculated along the line between the metal centres (Fig. S14, ESI†) exhibits smooth behaviour without indication of bonding for 1 and 5-Cl2, while there is an anomaly for 5 on the Pt(II) side signifying the tendency of the electrons to engage in bonding. The final indication that the Pt(II)–Rh(III) interaction in 5 is different from and stronger than its analogues in 1 and 5-Cl2 stems from the fact that the gas-phase DFT optimization leads to a metal–metal distance (2.715 Å) very close to the experimental X-ray values (2.6972(12) and 2.7172(13) Å in two crystallographically unrelated molecules containing ordered metal ions). For the remaining complexes, the differences are larger (1: 2.867(1) Å X-ray vs. 2.962 Å DFT; 5-Cl2: 2.911(6) Å X-ray vs. 2.995 Å DFT). Thus, the crystal packing forces are not decisive in shortening the metal–metal distance in 5, but rather a weak interaction is involved.
To conclude, a new synthetic arena for metal–metal interactions has been documented. By sacrificing two NH groups of the core of a classical porphyrin, we gained space to accommodate two transition metal ions nearly in the macrocyclic plane, in an atypical mutual orientation. The first heterobimetallic 21,23-dimetallaporphyrin, 21-platina-23-rhodaporphyrin 5, allowed for a weak metal–metal bond formation within a porphyrin core. The metal–metal interaction in 5 may be turned off by platinum(II) oxidation to platinum(IV) in 5-Cl2 and turned on in the relevant reduction.
Grzegorz Vetter: investigation (synthesis and spectroscopic studies), visualization, writing – original draft; Agata Białońska: formal analysis (crystallography); Aneta Jezierska & Jarosław J. Panek: formal analysis (calculations), writing – original draft; Ewa Pacholska-Dudziak: conceptualization, supervision, funding acquisition, visualization, writing – original draft, writing – review & editing.
We thank Professor Piotr J. Chmielewski for his help in electrochemical analysis. Financial support from the National Science Center (Grant 2020/37/B/ST4/00869) is kindly acknowledged. DFT calculations were carried out by using resources provided by the Wroclaw Centre for Networking and Supercomputing, Grant 329, and ACK Cyfronet Kraków Centre (ARES, part of the PL-Grid infrastructure).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Campos, Nat. Rev. Chem., 2020, 4, 696–702 CrossRef CAS PubMed.
- J. F. Berry and C. C. Lu, Inorg. Chem., 2017, 56, 7577–7581 CrossRef CAS PubMed.
- P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed.
- T. Nguyen, A. D. Sutton, M. Brynda, J. C. Fettinger, G. J. Long and P. P. Power, Science, 2005, 310, 844–847 CrossRef CAS PubMed.
- B. O. Roos, A. C. Borin and L. Gagliardi, Angew. Chem., Int. Ed., 2007, 46, 1469–1472 CrossRef CAS PubMed.
- L. Jin, D. R. Tolentino, M. Melaimi and G. Bertrand, Sci. Adv., 2015, 1, e1500304 CrossRef PubMed.
- J. Campos, J. Am. Chem. Soc., 2017, 139, 2944–2947 CrossRef CAS PubMed.
- M. Navarro, J. J. Moreno, M. Pérez-Jiménez and J. Campos, Chem. Commun., 2022, 58, 11220–11235 RSC.
- J. P. Collman and H. J. Arnold, Acc. Chem. Res., 1993, 26, 586–592 CrossRef CAS.
- A. B. Alemayehu, L. J. Mccormick-Mcpherson, J. Conradie and A. Ghosh, Inorg. Chem., 2021, 60, 8315–8321 CrossRef CAS PubMed.
- M. Toganoh, T. Niino, H. Maeda, B. Andrioletti and H. Furuta, Inorg. Chem., 2006, 45, 10428–10430 CrossRef CAS PubMed.
- J.-M. Barbe and R. Guilard, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, 2000, vol. 3, pp. 211–244 Search PubMed.
- M. J. Białek, P. J. Chmielewski and L. Latos-Grażyński, Chem. – Eur. J., 2019, 25, 14536–14545 CrossRef PubMed.
- T. Sarma, B. S. Kumar and P. K. Panda, Angew. Chem., Int. Ed., 2015, 54, 14835–14839 CrossRef CAS PubMed.
- G. Vetter, A. Białońska, P. Krzyszowska, S. Koniarz and E. Pacholska-Dudziak, Chem. – Eur. J., 2022, 28, e202201513 CrossRef CAS PubMed.
- E. Pacholska-Dudziak, L. Szterenberg and L. Latos-Grażyński, Chem. – Eur. J., 2011, 17, 3500–3511 CrossRef CAS PubMed.
- T. D. Lash, S. A. Jones and G. M. Ferrence, J. Am. Chem. Soc., 2010, 132, 12786–12787 CrossRef CAS PubMed.
- E. Pacholska, L. Latos-Grażyński and Z. Ciunik, Angew. Chem., Int. Ed., 2001, 40, 4466–4469 CrossRef CAS PubMed.
- B. Sathyamoorthy, A. Axelrod, V. Farwell, S. M. Bennett, B. D. Calitree, J. B. Benedict, D. K. Sukumaran and M. R. Detty, Organometallics, 2010, 29, 3431–3441 CrossRef CAS.
- E. Pacholska-Dudziak, M. Szczepaniak, A. Książek and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2013, 52, 8898–8903 CrossRef CAS PubMed.
- E. Pacholska-Dudziak, G. Vetter, A. Góratowska, A. Białońska and L. Latos-Grażyński, Chem. – Eur. J., 2020, 26, 16011–16018 CrossRef CAS PubMed.
- G. Vetter, A. Białońska and E. Pacholska-Dudziak, Inorg. Chem., 2023, 62, 3056–3066 CrossRef CAS PubMed.
- A. Srinivasan, M. Toganoh, T. Niino, A. Osuka and H. Furuta, Inorg. Chem., 2008, 47, 11305–11313 CrossRef CAS PubMed.
- N. Halder, K. C. Sahoo, K. Gourav, D. Usharani and H. Rath, J. Org. Chem., 2021, 86, 8015–8026 CrossRef CAS PubMed.
- B. M. Still, P. G. A. Kumar, J. R. Aldrich-Wright and W. S. Price, Chem. Soc. Rev., 2007, 36, 665–686 RSC.
- A. Gelling, K. G. Orrell, A. G. Osborne and V. Šik, J. Chem. Soc., Dalton Trans., 1998, 27, 937–945 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic, spectroscopic and theoretical analysis. CCDC 2241158 (5) and 2241159 (5-Cl2). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc01367g |
| This journal is © The Royal Society of Chemistry 2023 |