
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stable glycosylamines at the reducing ends of cellulose nanocrystals†
Jingwen
Xia
 a,
Tetyana
Koso
a,
Katja
Heise
b,
Lukas
Fliri
b,
Emilie
Ressouche
b,
Johanna
Majoinen
b,
Mauri A.
Kostiainen
b,
Sami
Hietala
a,
Michael
Hummel
b,
Vladimir
Aseyev
*a,
Ilkka
Kilpeläinen
*a and
Alistair W.T.
King
*c
a,
Tetyana
Koso
a,
Katja
Heise
b,
Lukas
Fliri
b,
Emilie
Ressouche
b,
Johanna
Majoinen
b,
Mauri A.
Kostiainen
b,
Sami
Hietala
a,
Michael
Hummel
b,
Vladimir
Aseyev
*a,
Ilkka
Kilpeläinen
*a and
Alistair W.T.
King
*c
aChemistry Department University of Helsinki, AI Virtasen Aukio 1, Helsinki FI-00560, Finland. E-mail: vladimir.aseyev@helsinki.fi; ilkka.kilpelainen@helsinki.fi
bDepartment of Bioproducts and Biosystems Aalto University Otakaari 1B, FI-00076, Espoo, Finland
cBA54 Biomaterial processing and products VTT Technical Research Centre of Finland Ltd Tietotie 4e, FI-02150, Espoo, Finland. E-mail: alistair.king@vtt.fi
First published on 4th July 2023
Abstract
The reaction of reducing end groups in cellulose nanocrystals with dodecylamine was examined. Using a direct-dissolution solution-state NMR protocol, the regioselective formation of glucosylamines was shown. This provides an elegant approach to sustainably functionalize these bio-based nanomaterials, that may not require further reduction to more stable secondary amines.
Regio-selective modifications of cellulose nanocrystals (CNCs) are experiencing increasing attention as an avenue toward designing functional nanomaterials; from liquid crystals1 to complex 2D nanostructures.2,3 Functionalization of the reducing end groups (REGs) of the cellulose chains, by means of aldehyde-specific chemistry, is probably the most prominent avenue, yielding anisotropically modified nanoparticles.4,5 Two synthetic approaches have been mainly explored, by targeting the reducing end ‘aldehydes’ directly with hydrazine derivatives (ligation reactions),6–9 or primary amines through reductive amination.10,11 A new approach using dicarbonyls12 (Knoevenagel condensation), may also show promise in the future. All these reactions are carried out under heterogeneous conditions, i.e., modifying the CNCs while dispersed in water, typically at a low consistency. To compensate for the high dilutions and to drive the reaction to completion, reagents are often used in excessive stoichiometries, at the cost of scalability and atom economy. Moreover, the chemistry applied to the reducing end of CNCs is often not well understood, due to the lack of suitable methods for structural characterisation. Recently, a solution-state NMR technique using the electrolyte [P4444][OAc]:DMSO-d6 to dissolve crystalline celluloses has emerged,13–15 enabling profound analysis at the molecular level. Combined with model reactions on water-soluble saccharides, this method allowed to confirm the selectivity of Knoevenagel condensation12 and reductive amination16 toward CNC REGs.
Given the commercial availability of different amines, reductive amination chemistry seems promising for CNC derivatization. However, there are drawbacks when upscaling. Most reports involve the use of significant excesses of reagents (e.g., 300 eq. reducing agent in a recent report16). Additionally, the use of typical borohydride reductants, e.g., sodium cyanoborohydride, causes contamination with residual boron-containing by-products. Common dialysis protocols needed to successfully tackle such contaminations of modified CNCs are not economical at scale. The same applies for azeotropic distillation, which was proposed for the removal of 2-picoline-borane (PICB)17 or 5-ethyl-2-picoline borane (PEMB).18 Another challenge emerges in identifying if the reduction step has even occurred. Even though several recent articles use reductive amination to modify the reducing ends of CNCs and plenty of indirect evidence shows the success of such reactions,10,11,19,20 only one article gives direct NMR spectroscopic evidence of secondary amine formation.1
In general, when targeting REGs for the selective modification of CNCs, the reactivity of the REG is presumed to be as the ‘aldehyde’ form, as in organic chemistry textbooks. However, the peculiar and complex chemistry of sugar aldehydes is well known in other research areas. The presence of several hydroxy functionalities in the structure influences the overall reactivity of the aldehyde over the presence of ring-chain equilibria or the potential occurrence of further rearrangements. Especially in investigations towards reactions of saccharides with amines these phenomena prove to be consequential. This is well documented in the Maillard reaction, where initial steps involve imine formation and subsequent rearrangements, or further cascade reactions, leading to a wide variety of products, generally associated with food aromas.21 These initial reactions are also important for the synthetic stabilization and bioavailability of proteins,22,23 with the first naturally occurring protein glycosylamine linkages identified in 1961.24 Aside from the synthesis of complex organic molecules, such chemistry has also been proven to be commercially relevant, e.g., in the production of surfactants via: (A) the reductive amination of sugars with fatty amines,25 or (B) the lipase-catalysed reaction of fatty acid carbonyls with N-methylglucamines, to form acylglucamides.26
Irrespective of the intended products the first stage in the reaction of sugar REGs with an amine is believed to involve the formation of an imine at the intermediate open-chain aldehyde of the terminal saccharide (Scheme 1). The carbonyl and imine species are in equilibrium with their respective anomeric cyclic structures. In the case of glucose, as an intensively studied model compound, these include its α and β anomers and the derivative α and β glucosylamines (Scheme 1A). In water and in the absence of reducing conditions, the bond between amine and sugar is known to be rather hydrolytically labile. However, the equilibria can be influenced by the amine substituents, e.g., over their electron donating or withdrawing nature, their steric demand and hydrophobicity. Also, solvent effects and pH of the media are important factors.27
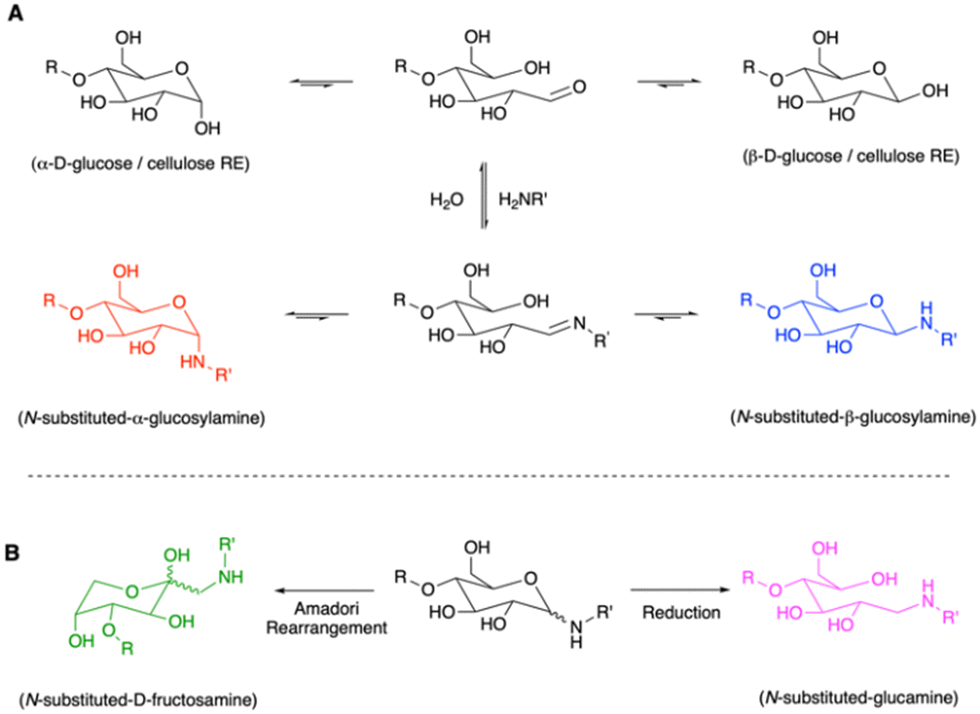 | ||
| Scheme 1 Known pathways involved in the reaction of glucose with primary amines - probed for the modification of the REGs of CNCs in this work: (A) In contact with solvent the more stable α and β anomers of the reducing sugars are in equilibrium with their open chain form. In the presence of an amine the aldehyde can react under formation of an imine (Schiff base), which is in turn in an equilibrium with the α and β glucosylamines; (B) Potential side reactions during glucosylamine preparation are Amadori rearrangements, resulting in fructosamines. These represent intermediates in the Maillard reaction cascade. The reductive amination of CNCs to glucamines can circumvent this and potential hydrolytic stability issues but at cost. (R = H for glucose; R = anhydroglucose units (AGUs) for cellulose; R′ = alkyl). | ||
While rapid hydrolysis can be expected in many cases, certain N-substituted glucosylamines and other glycosylamines (derived from other monosaccharides than glucose) are stable, isolable, and well-studied, especially concerning glycoconjugation, further Amadori rearrangements28 and Maillard cascade reactions.21 Many synthetic procedures exist for isolation of glycosylamines. They are reported to become more hydrolytically stable at higher pH ranges,27 with the Amadori rearrangement (Scheme 1B) promoted under addition of catalytic amounts of weak acids.29–31
In the case of nanocellulose modification, as mentioned before, excess reducing agent and amine is typically applied to drive the reaction to completion, to yield a hydrolytically stable N-substituted glucamine terminus (i.e., secondary amine, Scheme 1B). Considering the discussed drawbacks, we were interested if REG modifications with primary amines can also be performed in a more sustainable and scalable manner. While this represents a rather trivial challenge from an organic chemist's preparative perspective – apart from the heterogenous reaction conditions needed for CNC modification – the unambiguous identification of the planned chemistry remains complex. However, understanding of the formed structures is important as they strongly influence the stability of the derivatization and, thus, set the scope for potential applications. If we wish to move toward sustainably modified cellulosics and more specifically nanocellulose REG modifications, we need to begin to chart the chemistry at the REG – by providing direct high-resolution spectroscopic proof, as is standard in organic chemistry.4
Moreover, scalable, economically, and ecologically viable alternatives for CNC modifications must be sought, where the mechanistic understanding is the critical starting point, to avoid the existing problems. In this sense we showcase, herein, how formation of glycosylamines, at the REGs of CNCs, can be elucidated by applying a developing solution state NMR protocol utilising direct dissolution of the CNCs into an ionic liquid-DMSO-d6 electrolyte.13–15
After a preliminary screening study (ESI,† S2), reaction of glucose with the long-chain aliphatic primary amine dodecylamine (DDA) gave a stable product for further analysis. Consequently, a model glucosylamine (anomeric mixture of α and β) was prepared by reaction of glucose with DDA (1.2 eq. to glucose). Recrystallization of the α and β mixture resulted in the isolation of a sample of pure β glucosylamine, for 1H-13C HSQC NMR spectral assignment. The assignments for the β anomer and the α and β mixture are shown in ESI,† Fig. S2. For comparison, the reductive amination of glucose was performed by addition of PEMB to yield the dodecylglucamine for HSQC NMR signal assignment (ESI,† Fig. S3). Clearly there is a difference between the 1- and 2-position saccharide resonances in the 1H and HSQC spectra for the dodecylglucamine – the main diagnostic peaks in the HSQC spectrum (the anomeric CH-1 correlations) are missing in the dodecylglucamine sample. The gem-α correlations are also now shifted downfield in both spectral ranges.
Given the successful preparation of the model compound we applied the reaction conditions (0.24 eq. of DDA vs AGU, heating at 70 °C for 3 d) to commercial CNCs. To examine solvent effects the reactions were performed both as dispersions in water and anhydrous DMSO. It was assumed that the reaction in DMSO would be a lot more amenable to shifting the equilibrium towards the product and allow for easier access to the reducing ends, due to lower aggregation of the CNC dispersion in dipolar aprotic solvents. The products were analysed by diffusion-edited 1H NMR (ESI,† Fig. S4), in the [P4444][OAc]:DMSO-d6 electrolyte (direct dissolution NMR solvent),13,14 which clearly indicated that the reaction had occurred in both solvents. Despite strong peak superposition with the used electrolyte in other NMR experiments, the diffusion-editing allows to straightforwardly examine the derivatization. The editing removes the fast-diffusing species, e.g. H2O, [P4444][OAc], DMSO-d6 and DDA, leaving only the slow-diffusing polymeric species (including DDA covalently attached to the CNCs) visible in the spectra. Expectedly, the reaction in DMSO showed a higher conversion to the aminated product, evident by the increase in intensity of the aliphatic signals (<3 ppm), relative to the water reaction. At this point it is unclear if this is due to decreased aggregation giving access to REGs (in DMSO) vs. equilibrium shift in the presence of H2O, or some additional kinetic effect. However, it is clear that the aminated products, in the absence of reducing agent, are relatively stable in both DMSO and H2O.
The DDA-CNC glycosylamine material prepared in DMSO was further analysed using multiplicity-edited HSQC, in [P4444][OAc]:DMSO-d6, using a higher field 850 MHz 1H frequency spectrometer equipped with a N2-cooled cryoprobe. As can be seen from the HSQC spectrum (Fig. 1), both glucosylamine end-group anomers as identified in the glucose model compounds (ESI,† Fig. S2) are present. Moreover, they are in the correct proportions (allowing for HSQC quantification error), relative to the α and β glucosylamine model mixture and are in excess over the residual REGs of cellulose. The reaction has not gone to completion, with a healthy ∼80% conversion based on the relative intensities in Fig. 1. Considering the heterogenous reaction conditions this represents an acceptable yield. One can speculate that aggregation or sterics play a role in the non-quantitative transformation, in addition to the potential for slow hydrolysis during workup. Noteworthy, there were also no apparent signs for further follow-up reactions, conceivably following Amadori rearrangements or even the Maillard reaction cascade. This proves that the formed glucosylamine structures in the DDA-CNC derivatives are stable towards work-up and dissolution in the NMR electrolyte over prolonged times, at 65 °C. Another aspect that can affect redispersibility and reactivity of the CNCs is the presence of sulphate half esters on the CNC surface. These do not seem to be removed or converted to dodecylsulphonamide, due to the visible retention of the 6-OH sulphate half ester functionalities (gem-6-OSO3−) in the HSQC spectrum (Fig. 1).
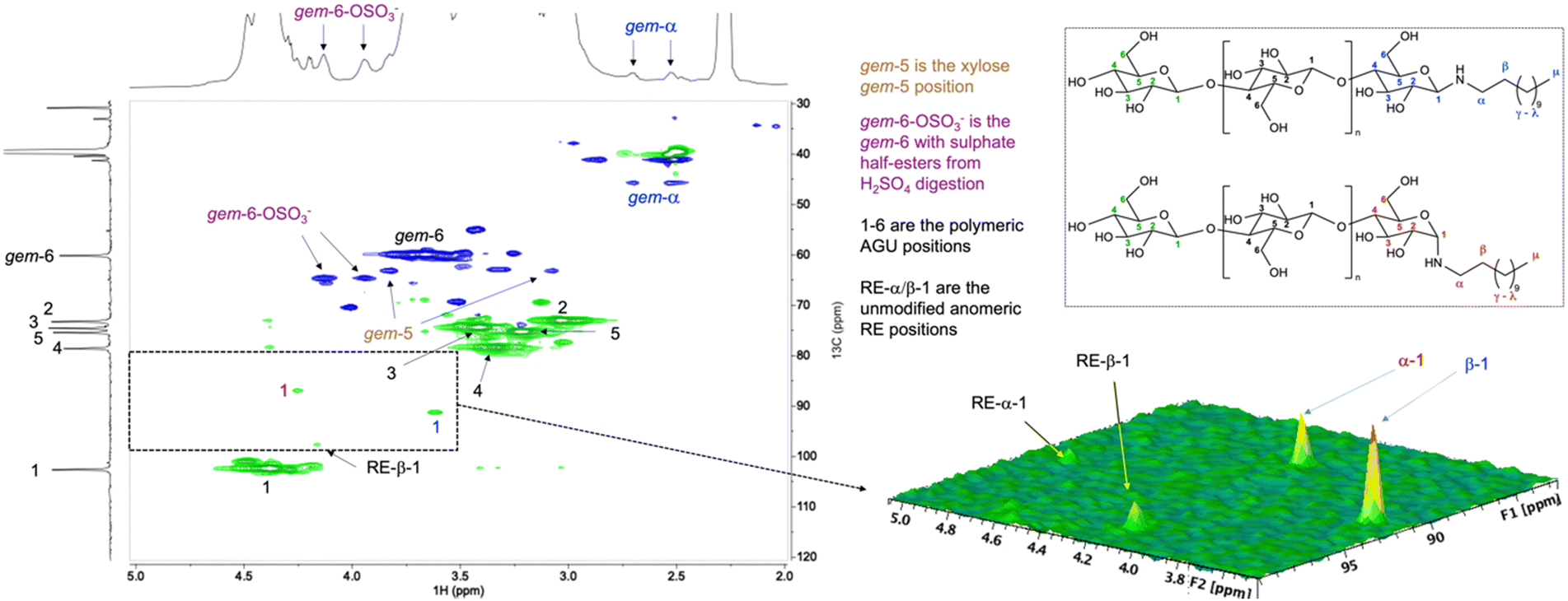 | ||
Fig. 1 Selective modification of the REGs with DDA applied to a commercial sulphated CNC. This is a more complex cellulosic substrate with relevance in materials science. (a) The multiplicity-edited 1H-13C HSQC spectrum ([P4444][OAc]:DMSO-d6 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 wt%), 850 MHz, 65 °C, 5 wt% CNC) showed signals characteristic for the α and β glucosylamine structures (see Scheme 1). (b) 3D expansion of the C1-H spectral area demonstrating a conversion of around 80%, based on the relative intensities of the REG and glucosylamine anomeric signals –CH and CH3 groups are shown in green, CH2 groups are shown in blue. Diffusion-edited 1H trace shown on top and 13C trace on the left. :4 wt%), 850 MHz, 65 °C, 5 wt% CNC) showed signals characteristic for the α and β glucosylamine structures (see Scheme 1). (b) 3D expansion of the C1-H spectral area demonstrating a conversion of around 80%, based on the relative intensities of the REG and glucosylamine anomeric signals –CH and CH3 groups are shown in green, CH2 groups are shown in blue. Diffusion-edited 1H trace shown on top and 13C trace on the left. | ||
For comparison, the reductive amination of the commercial CNCs was also tested using DDA and PEMB as reducing agent. An HSQC spectrum was also obtained using the more sensitive equipment (ESI,† Fig. S5). By comparison with our glucamine and glucosylamine models, it was clear that the reductive amination had not occurred, by the absence of corresponding glucamine correlations. Moreover, glycosylamine resonances were also missing, as were the resonances corresponding to the REGs. This tentatively indicates that reduction of the REGs seems to occur much faster than reduction of the Schiff base.
While the above analyses all demonstrate the stability of dodecyl-N-glycosylamine conjugates, with glucose and CNCs, their presence in the solid-state is still under question; all previous and literature spectra demonstrate stability only in the solution-state. As such, the 13C solution-state NMR spectrum of the α and β mixture of the dodecylglucamine was compared with the solid-state 13C spectrum, utilizing cross-polarization (CP) and magic-angle spinning (MAS) (ESI,† Fig. S6). This demonstrates the stability of the glucosylamine moiety in both the solid-state, and in the solution-state, including under ambient neutral aqueous conditions.
As such, the modification of the REGs of CNCs is clearly possible, in the absence of reducing reagents, yielding a water-stable glycosylamine product, dependent on the starting amine. This brings the application of REG-functionalized cellulosics a step closer to reality, particularly in the areas of materials and biomedical sciences.
One should also now consider that indirect proof of REG reductive aminations, using current protocols, cannot exclude the presence of hydrolytically unstable non-reduced glycosylamine structures. For the current approach, hydrolytic stability of different REG substituents now needs to be charted, as a function of the differing aqueous conditions, the electron-withdrawing nature, and the hydrophobic nature of the substituents. Reaction optimization and the effect of aggregation of CNCs under ‘green’ processing conditions clearly also should be probed. It is important to consider that the presented derivatization approach might not be applicable to a wider variety of primary amines, owing to rearrangement side reactions. This was suggested by the brown coloration of mixtures when employing short chain aliphatic amines, in our pre-screening experiments, hinting towards the occurrence of Maillard reactions (also known as ‘browning’ reactions). However, this may also provide another angle on derivatization strategies over the isolation of the intermediately formed and more stable Amadori rearrangement product, which may avoid potential hydrolysis over wider pH conditions. There are a plethora of reports exploiting the peculiar properties of sugar REGs for conjugation reactions. Arguably, adapting these protocols for cellulose modification, after fine tuning the reaction conditions using suitable analytical tools, represents a more elegant derivatization approach than employing super-stoichiometric amounts of potentially toxic co-reagents. Overall, we hope that this work demonstrates that it is possible to elevate cellulose chemical optimization studies to a more fundamental bottom-up approach, by applying similar analytical methodology to that which has been applied in organic chemistry since the 1960's, i.e., solution-state NMR.
The authors wish to acknowledge the Research Council of Finland for funding (Project's: 310481 & 311255). The facilities and expertise of the HiLIFE NMR unit at the University of Helsinki, a member of Instruct-ERIC Centre Finland, FINStruct, and Biocenter Finland are gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Delepierre, H. Traeger, J. Adamcik, E. D. Cranston, C. Weder and J. O. Zoppe, Biomacromolecules, 2021, 22, 3552 CrossRef CAS PubMed.
- F. Lin, F. Cousin, J.-L. Putaux and B. Jean, ACS Macro Lett., 2019, 8, 345 CrossRef CAS PubMed.
- A. Villares, C. Moreau and B. Cathala, ACS Omega, 2018, 3, 16203 CrossRef CAS PubMed.
- K. Heise, G. Delepierre, A. W. T. King, M. A. Kostiainen, J. Zoppe, C. Weder and E. Kontturi, Angew. Chem., Int. Ed., 2021, 60, 66 CrossRef CAS PubMed.
- H. Tao, N. Lavoine, F. Jiang, J. Tang and N. Lin, Nanoscale Horiz., 2020, 5, 607 RSC.
- J.-L. Huang, C.-J. Li and D. G. Gray, ACS Sustainable Chem. Eng., 2013, 1, 1160 CrossRef CAS.
- E. Sipahi-Sağlam, M. Gelbrich and E. Gruber, Cellulose, 2003, 10, 237 CrossRef.
- H. Sadeghifar, I. Filpponen, S. P. Clarke, D. F. Brougham and D. S. Argyropoulos, J. Mater. Sci., 2011, 46, 7344 CrossRef CAS.
- M. A. Karaaslan, G. Gao and J. F. Kadla, Cellulose, 2013, 20, 2655 CrossRef CAS.
- L. R. Arcot, M. Lundahl, O. J. Rojas and J. Laine, Cellulose, 2014, 21, 4209 CrossRef CAS.
- A. R. Lokanathan, A. Nykänen, J. Seitsonen, L.-S. Johansson, J. Campbell, O. J. Rojas, O. Ikkala and J. Laine, Biomacromolecules, 2013, 14, 2807 CrossRef CAS PubMed.
- K. Heise, T. Koso, L. Pitkänen, A. Potthast, A. W. T. King, M. A. Kostiainen and E. Kontturi, ACS Macro Lett., 2019, 8, 1642 CrossRef CAS PubMed.
- L. Fliri, K. Heise, T. Koso, A. R. Todorov, D. Rico del Cerro, S. Hietala, J. Fiskari, I. Kilpeläinen, M. Hummel and A. W. T. King, Nat. Protoc., 2023, 18, 2084 CrossRef PubMed.
- A. W. T. King, V. Mäkelä, S. A. Kedzior, T. Laaksonen, G. J. Partl, S. Heikkinen, H. Koskela, H. A. Heikkinen, A. J. Holding, E. D. Cranston and I. Kilpeläinen, Biomacromolecules, 2018, 19, 2708 CrossRef CAS PubMed.
- T. Koso, D. Rico del Cerro, S. Heikkinen, T. Nypelö, J. Buffiere, J. E. Perea-Buceta, A. Potthast, T. Rosenau, H. Heikkinen, H. Maaheimo, A. Isogai, I. Kilpeläinen and A. W. T. King, Cellulose, 2020, 27, 7929 CrossRef CAS.
- G. Delepierre, K. Heise, K. Malinen, T. Koso, L. Pitkänen, E. D. Cranston, I. Kilpeläinen, M. A. Kostiainen, E. Kontturi, C. Weder, J. O. Zoppe and A. W. T. King, Biomacromolecules, 2021, 22, 2702 CrossRef CAS PubMed.
- E. R. Burkhardt, Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2010 Search PubMed.
- E. R. Burkhardt and I. Sharma, Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2010 Search PubMed.
- L. Li, H. Tao, B. Wu, G. Zhu, K. Li and N. Lin, ACS Sustainable Chem. Eng., 2018, 6, 14888 CrossRef CAS.
- H. Tao, A. Dufresne and N. Lin, Macromolecules, 2019, 52, 5894 CrossRef CAS.
- J. E. Hodge, J. Agric. Food Chem., 1953, 15, 928 CrossRef.
- R. J. Solá and K. Griebenow, J. Pharm. Sci., 2009, 98, 1223 CrossRef PubMed.
- S. V. Moradi, W. M. Hussein, P. Varamini, P. Simerska and I. Toth, Chem. Sci., 2016, 7, 2492 RSC.
- P. G. Johansen, R. D. Marshall and A. Neuberger, Biochem. J., 1961, 78, 518 CrossRef CAS PubMed.
- K. Hill and O. Rhode, Eur. J. Lipid Sci. Technol., 1999, 101, 25 CAS.
- T. Maugard, M. Remaud-Simeon, D. Petre and P. Monsan, Tetrahedron, 1997, 53, 7629 CrossRef CAS.
- K. Villadsen, M. C. Martos-Maldonado, K. J. Jensen and M. B. Thygesen, ChemBioChem, 2017, 18, 574 CrossRef CAS PubMed.
- V. V. Mossine and T. P. Mawhinney, Adv. Carbohydr. Chem. Biochem., 2010, 64, 291 CrossRef CAS PubMed.
- H. S. Isbell and H. L. Frush, J. Org. Chem., 1958, 23, 1309 CrossRef CAS.
- M. Monsigny, C. Quétard, S. Bourgerie, D. Delay, C. Pichon, P. Midoux, R. Mayer and A. C. Roche, Biochimie, 1998, 80, 99 CrossRef CAS PubMed.
- J. E. Hodge, Adv. Carbohydr. Chem., 1955, 10, 169–205 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc01329d |
| This journal is © The Royal Society of Chemistry 2023 |