
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne reaction – double stereoinduction: C–H activation as a privileged route towards complex atropisomeric molecules
Amandine
Luc
and
Joanna
Wencel-Delord
 *
*
Laboratoire d’Innovation Moléculaire et Applications (UMR CNRS 7042), Université de Strasbourg/Université de Haute Alsace, ECPM. 25 rue Becquerel, 67087, Strasbourg, France. E-mail: wenceldelord@unistra.fr
First published on 14th June 2023
Abstract
Expanding the importance of chirality and implementation of stereogenic information within complex molecular design has recently reached a new level: design of innovative enantiopure scaffolds bearing multiple chiral elements. In particular, regarding sustainability aspects and straightforward use of relatively simple substrates, the C–H activation strategy offers unique opportunities to assemble complex chiral molecules with unique topologies while controlling two stereoselective events in a single transformation. Herein, the emerging field of asymmetric C–H activation allowing rapid construction of atropisomeric molecules bearing a second chirality element, such as a stereogenic center, vicinal chiral axis or planar chirality, is described. Aiming at in-depth comprehension of such innovative systems, the emphasis is put on the nature of stereodiscriminant steps, allowing the simultaneous control of both chiral elements.
Introduction
With the expanding importance and quest for enantiopure molecules, asymmetric synthesis has been one of the most rapidly developing fields of organic synthesis. Indeed, while the end of the 20th century witnessed a growing interest in enantiopure drugs featuring improved reactivities, selectivities, and fewer side effects,1 the beginning of the 21st century is clearly marked by the implementation of chirality in various fields of chemistry, including agrochemistry, material science, catalyst design, etc.2 The expanding importance and applications of enantiopure molecules clearly translate into a need for more efficient and sustainable synthetic routes to prepare such compounds on one hand, and the design of original chiral scaffolds with appealing applications on the other. For decades, central chirality arising from the C-stereogenic atom has been the most common and the most investigated stereogenic element. However, over the last decade, other types of chiral elements, including planar chirality or atropoisomerism resulting from a restricted rotation around Ar–Ar, Ar–N, or Ar–styrene bonds have spread (Scheme 1a). Indeed, such complementary stereoelements give a unique opportunity to incorporate new properties within a compound and escape from flatness. To further explore and expand the field, over the last few years, growing attention has also been devoted to the design and synthesis of even more complex chiral molecules, bearing multiple chirality elements.3 In particular, the design of synthetic routes allowing the simultaneous control of two chiral events within one step has become an appealing synthetic challenge.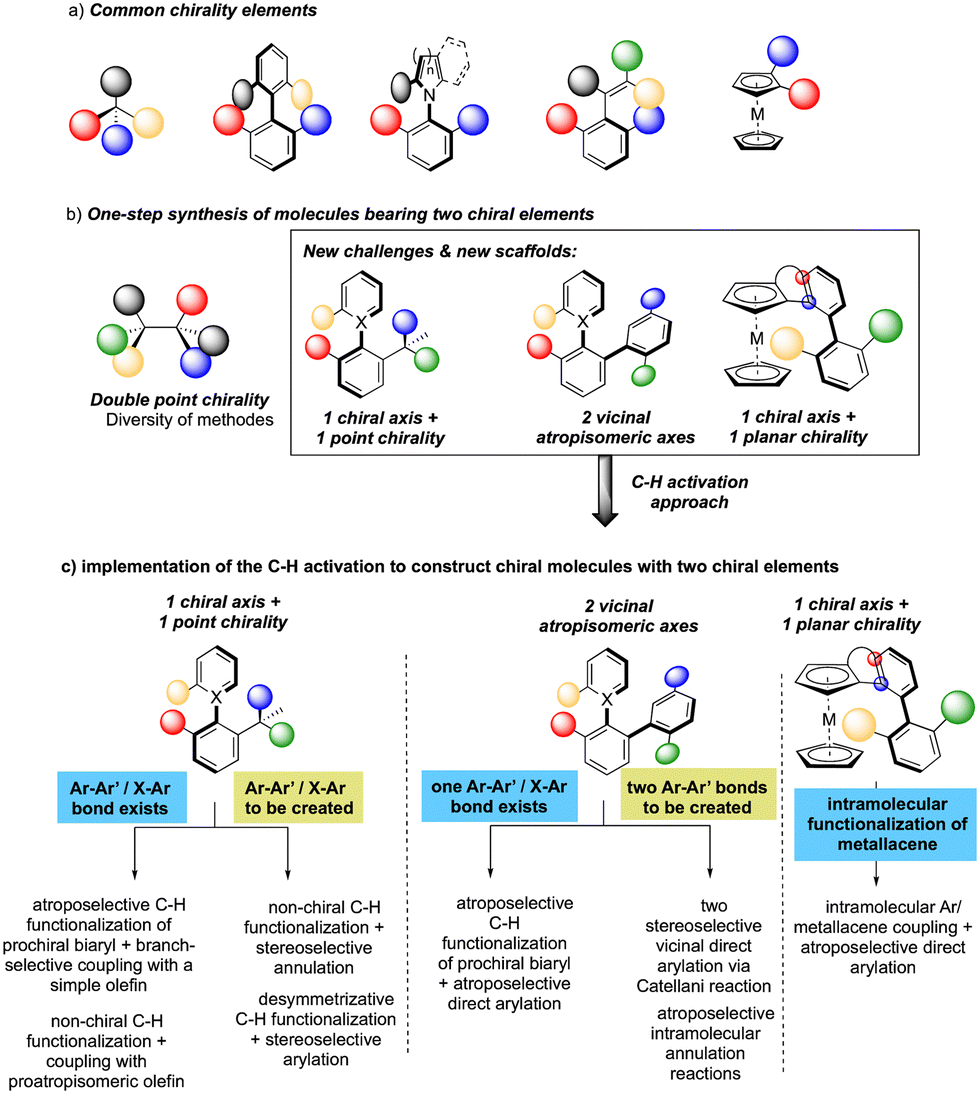 | ||
| Scheme 1 (a) Different types of chirality; (b) various types of multi-chiral molecules; (c) this work: C–H activation-based protocols to assemble atropisomeric compounds bearing an additional chirality element. | ||
While one-step synthesis of molecules bearing two C-stereogenic carbon-centers may be easily achieved through well-known approaches such as functionalization of olefins, asymmetric hydrogenation, etc., simultaneous control of two atropisomeric elements or one chiral axis and one point chirality appears as a daunting task (Scheme 1b). And yet, the implementation of multichirality within complex drug-candidates has been attracting growing attention and is expected to englobe also other advanced molecules’ design.4
The extraordinary expansion of the C–H activation field over the last two decades may be clearly correlated with the urgent need for green and sustainable organic synthesis, providing access to molecular complexity while minimizing step numbers and waste generation.5 In parallel, the C–H activation strategy provides a unique possibility to design new retrosynthetic disconnections and rapidly access molecules difficult to assemble using well-established synthetic tools.6 The possibilities offered by C–H activation routes have been progressively embraced within the asymmetric synthesis, thus furnishing compounds exhibiting not only point chirality but also various atropisomeric elements.7 Remarkably, very recently C–H activation-based reaction also paved the way for the one-step construction of multi-chiral scaffolds. In this feature article, the implementation of C–H activation to access unique molecules showing either two atropisomeric elements or one chiral axis and one point-chirality is discussed (Scheme 1c). Our aim is to present the astute synthetic solutions and precise reactions’ design to implement and benefit from the direct C–H activation step to simultaneously induce both chiral elements. Of note is that although C–H activation/annulation-type reactions with olefins furnish products with two C-stereogenic centers, these reactions will not be discussed herein.
C–H activation strategy to simultaneously control axial and point chiralities
While considering the diversity of transformations allowing C(sp2)–H bond activation, it is not surprising that the implementation of this synthetic route to prepare atropisomeric compounds has attracted scientific curiosity as early as 2000, with the seminal contribution of Murai.8 Over the years, three complementary approaches9 have been established to assemble atropisomeric biaryls via C–H activation: (1) direct functionalization of atropounstable or prochiral biaryls; (2) direct stereoselective arylation and (3) coupling with cyclic diazo-type partners followed by rearomatization. While stereoselective functionalization of biaryls and heterobiaryls turned out to be a very general, versatile, and reliable route, atroposelective direct arylation is clearly more challenging.9a Indeed, atroposelective direct Ar–H/Ar–X couplings between two sterically hindered compounds usually require high reaction temperatures that might compromise the atropostability of the newly generated compounds under the reaction conditions. Initial efforts on doubly stereoselective C–H activation reactions concerned the desymmetrization of biaryls with concomitant control of the point chirality. Capitalizing on the extensive knowledge of C–H desymmetrization reactions using 2-phenylpyridine type compounds,8,10 Fernández, López-Serrano, Ros, Lassaletta, and coworkers surmised that direct hydroarylation of such compounds should provide the original chiral molecules with both axial and point-chirality, provided that branch-selectivity can be efficiently controlled.11 Design of a catalytic system combining Ir-precatalyst and spiro-diphosphine ligand L1 thus allowed efficient hydroarylation of various acyl vinyl ethers 2 and norbornenes using phenyl-pyridine and naphthyl-pyridine type substrates 1 (Scheme 2). The reaction occurs via dynamic kinetic resolution, i.e. the chiral catalyst reacts with both atropisomers of the substrate and the metallation/migratory insertion of the olefin sequence delivers selectively one atropisomer of the product 3 with not only a perfect control of branch vs. linear selectivity but also excellent diastereo- and enantio-selectivities. Experimental mechanistic studies combined with DFT calculations suggest the Chalk–Harrod-type mechanism to be operating with the migratory insertion of the olefin into the Ir–C bond of the atropisomeric metalacyclic intermediate being the selectivity-determining step.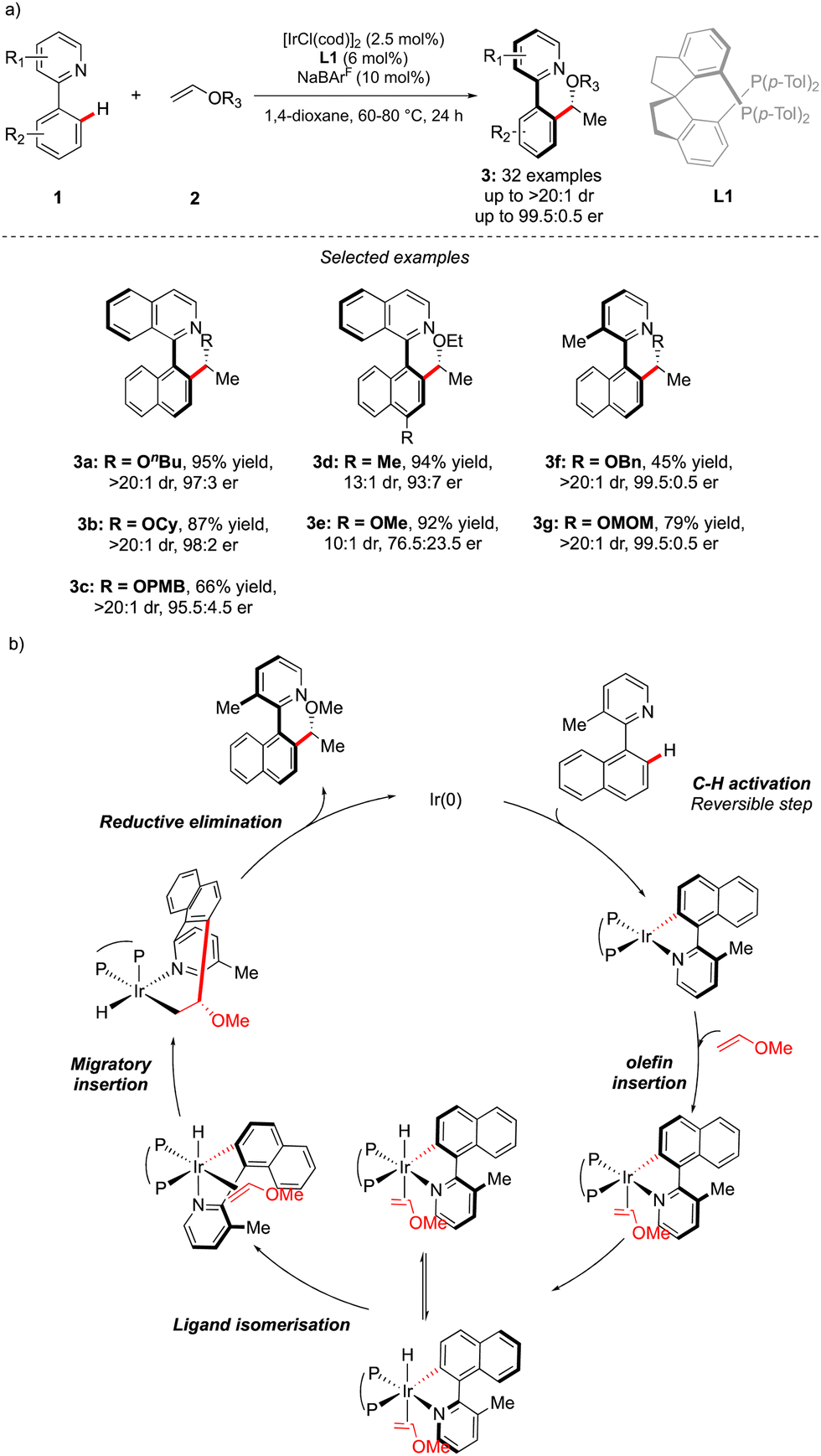 | ||
| Scheme 2 Synthesis of atropisomeric biaryls bearing an additional point chirality. | ||
Although out of scope as a metal-free transformation, worth mentioning is also a conceptually quite similar photocatalytic process, recently orchestrated by Xiao.12 In this case, precisely designed atropounstable 2-methyl-5-arylpyrimidine derivatives 4 undergo photo-induced Minisci reaction with N-acetyl α-amino-acid derivatives 5, furnishing ortho-alkylated atropisomeric products 6 (Scheme 3). Interestingly, the excellent induction of both, axial and central-chiralities is reached using enantiopure chiral phosphoric acid-type ligand L2.13
 | ||
| Scheme 3 Photocatalytic atroposelective Minisci-type reaction. | ||
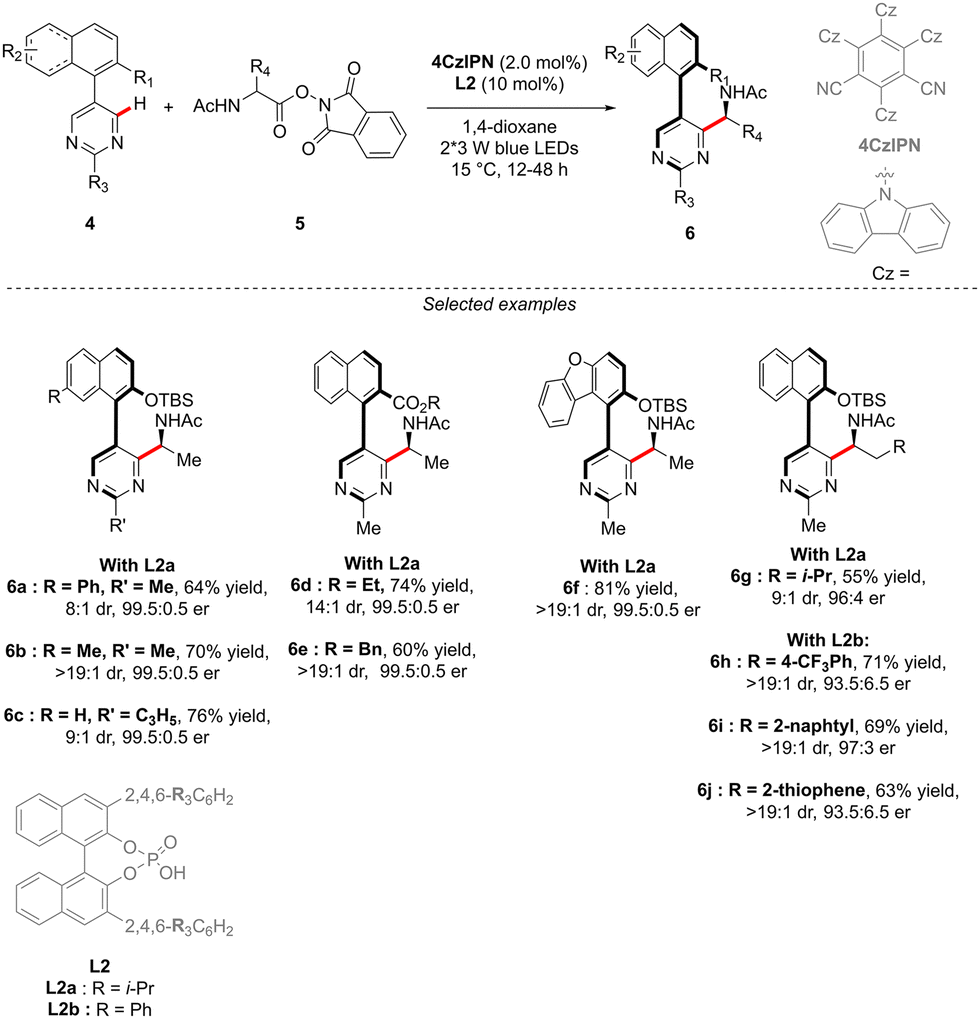
The concept reported by Fernández, López-Serrano, Ros, Lassaletta, and coworkers is not only limited to the synthesis of atropisomeric biaryls bearing an additional central chirality. Very recently, a similar methodology allowed the group of Ackermann to access the original C–N axially chiral scaffold showing an additional point-chirality (Scheme 4).14 In this case, Ru-catalyzed direct, branch-selective C2-alkylation of N-isoquinoline indoles 7 with various olefins 2 and vinylsilanes 8 was performed in both a diastereo- and enantioselective manner (Scheme 4). This unique transformation, benefiting from the directing group-ability of the pyridine motif, delivers a diversity of complex indoles with perfect control of both chiral elements. The efficient chirality transfer is reached using a common [RuCl2(p-cymene)]2 precatalyst in combination with a chiral carboxylic acid L3, implicitly participating in the reversible carboxylate-assisted C–H bond cleavage, thus imposing atropochirality. The chiral ligand contributes further to the chirality transfer via non-covalent interactions between the allylbenzene coupling partner and thiophene motif, hence enhancing the migratory insertion of the olefin into the Ru–C bond, followed by proto-demetalation with the chiral ligand, affording the desired product.
 | ||
| Scheme 4 Synthesis of C–N atropisomeric compounds with additional central chirality via atroposelective functionalization of a prochiral C–H substrate. | ||
In the above-introduced examples, the axial chirality and the central-chirality are typically introduced during two different elementary steps, i.e. enantioselective generation of the atropostable metalacyclic intermediate followed by diastereoselective migratory insertion. An alternative strategy to reach multi-chiral compounds implies a transformation during which both chirality elements are simultaneously set within one fundamental event of a catalytic cycle. Such a concept was elegantly illustrated in the recent work of Li reporting diastereo- and enantio-selective synthesis of C–N axially chiral benzoylpyrrolidine derivatives 12 (Scheme 5).15 In this work, and contrary to the previous examples, it is the finely designed olefin coupling partner that produces, upon the hydroarylation step, both axial and point-chiralities. The challenge of such a reaction design arises therefore from a relatively long distance between the chiral catalyst and the pro-atropisomeric element. Despite this strain, the desired highly enantio- and diastereoselective C–H alkylation of a simple C–H activation substrate: benzamide 10, using benzoylpyrrolidine 11 as the coupling partner was efficiently catalyzed by a chiral RhCp-derived catalyst C1. Under the optimized reaction conditions, essentially only one diastereoisomer of product 12 was obtained (d.r. > 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in high yields, regardless of the electronic properties of the benzamide substrate. The reaction tolerates well also various substituents on the pro-axially chiral N-arylmaleimides 11. Of note is that single-step atropo- and point-chirality induction may be rationalized based on Cramer's stereomodel, and the key repulsion between the most sterically hindered maleimide face and either the relatively bulky amide directing group or the tBu-substituent on the Cramer's Cp*-derived catalyst C1.
:1) in high yields, regardless of the electronic properties of the benzamide substrate. The reaction tolerates well also various substituents on the pro-axially chiral N-arylmaleimides 11. Of note is that single-step atropo- and point-chirality induction may be rationalized based on Cramer's stereomodel, and the key repulsion between the most sterically hindered maleimide face and either the relatively bulky amide directing group or the tBu-substituent on the Cramer's Cp*-derived catalyst C1.
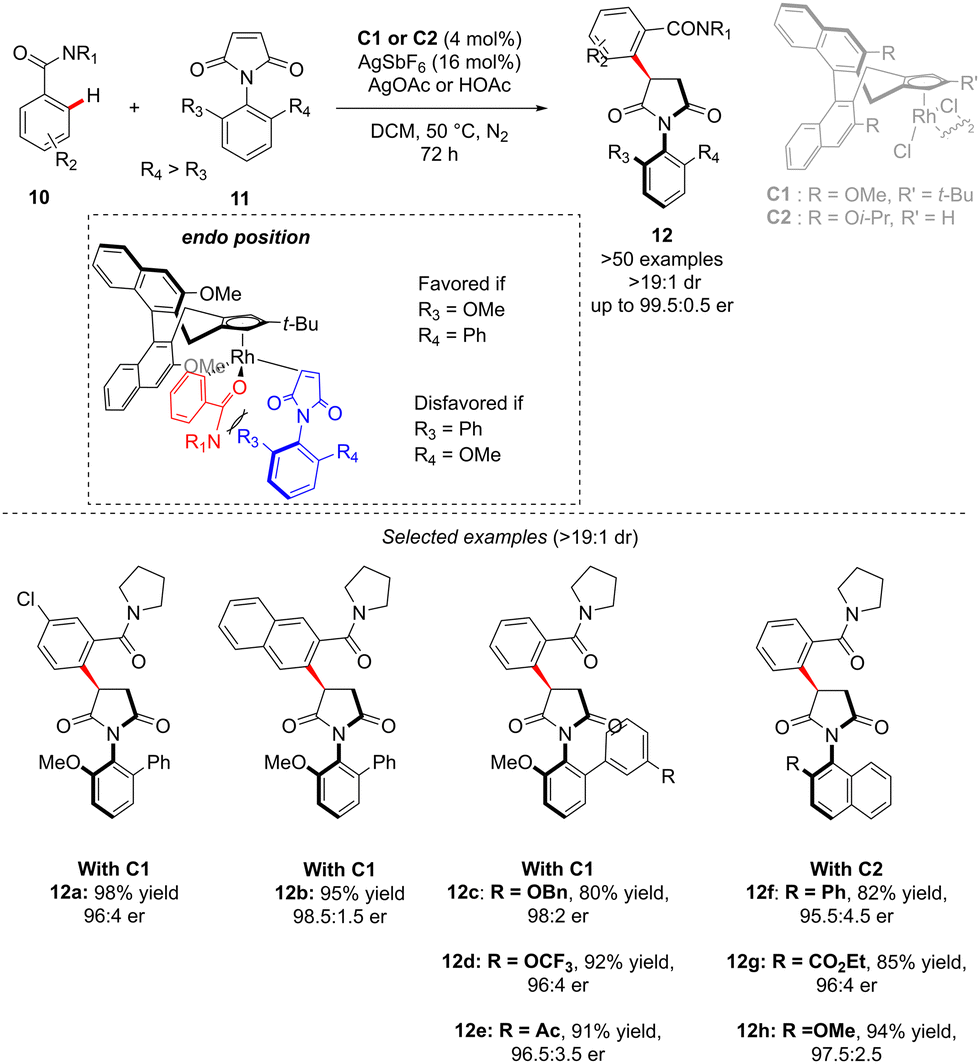 | ||
| Scheme 5 C–N + central chirality via non-chiral C–H activation followed by coupling with olefins bearing a pro-atropochiral motif. | ||
The generation of the atropisomeric element via C–H activation is not always limited to the astute functionalization of the already existing biaryl or N-aryl motifs. Although rare, the synthesis of atropisomeric compounds may also occur via stereoselective construction of the Ar–Ar′ bond as shown recently by Li and co-workers (Scheme 6).16 In this challenging project, atropoisomerism and C-central chirality were induced during the C–H activation/[3+2] annulation sequence. Nitrone 13 is indeed prompt to undergo metallation in the presence of chiral Rh-catalyst C3, followed by migratory insertion and annulation with alkynes. The precise design of the alkyne coupling partners 14, bearing an ortho-substituted naphthyl motif, gives therefore a possibility to build, via a [3+2] annulation step, unique axially and point-chiral indenes 15 under mild reaction conditions. Furthermore, the installation of the indole motif on the alkyne provides an interesting opportunity to afford C–N axially chiral indenones 15i–k. The DG-alkyne interactions are crucial for efficient stereocontrol and thus the desired products could be isolated in full diastereoselectivities (>15:1) and excellent enantioselectivities (typically er > 97.5:2.5). The mechanistic studies reveal that C–H activation is the rate-determining step, while the alkyne insertion is enantio-determining.
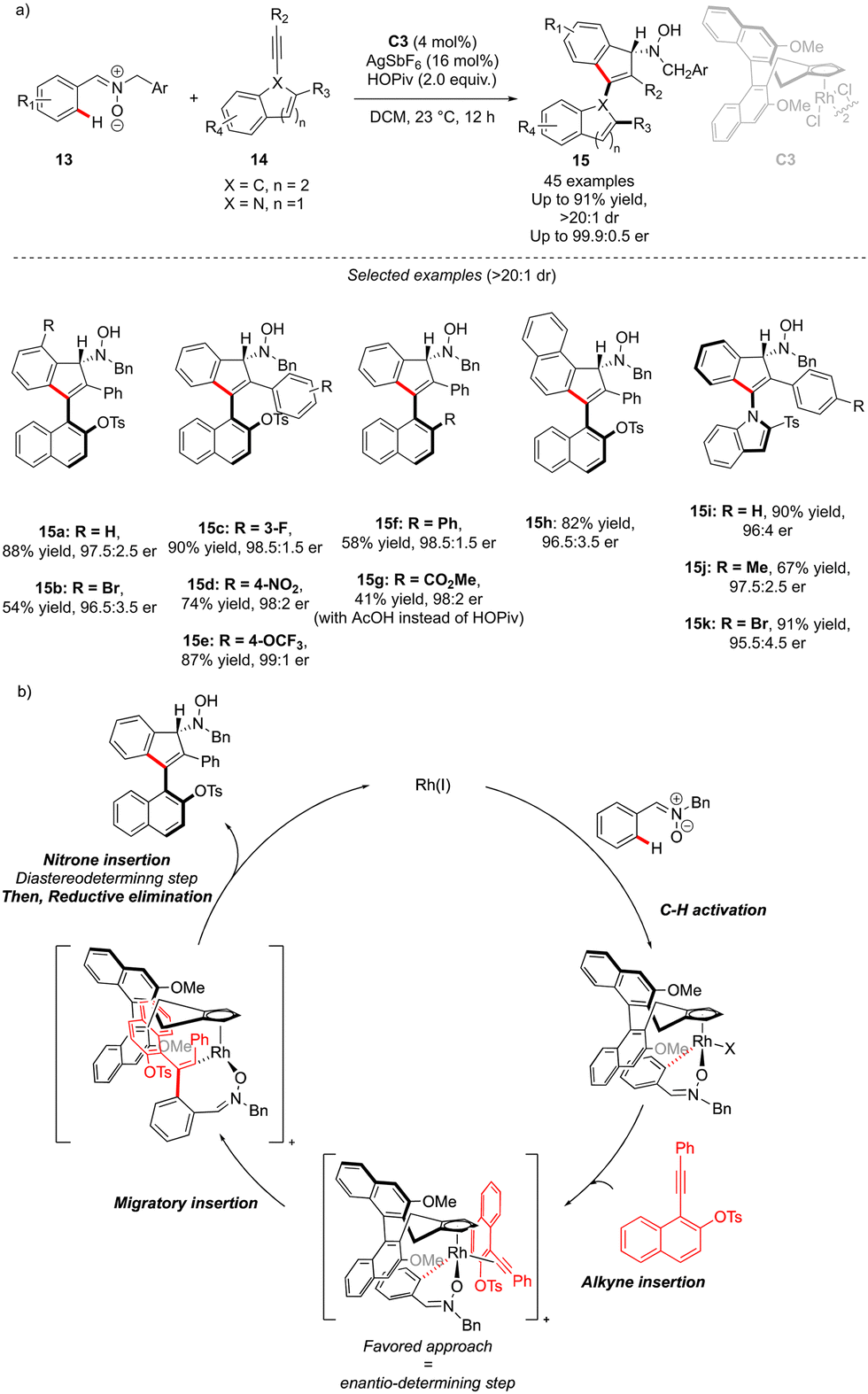 | ||
| Scheme 6 Synthesis of C–C or C–N atropisomeric compounds with additional point-chirality via construction of the aromatic ring. | ||
Although the most common, the central-chirality does not necessarily arise from the presence of a C-stereogenic centre and alternative motifs, such as stereogenic P-, Si- or S-atoms have been catching growing scientific curiosity.17 The group of Cramer focused therefore on the synthesis of original P- and axially-chiral products by implementing a C–H activation mindset. The pioneering efforts towards this goal, reported in 2018, focused on the desymmetrizative C–H arylation of diaryl-phosphine oxides 16 with o-quinone diazides 17 (Scheme 7).18 During such a transformation, not only is a P-stereogenic centre generated, but also the chirality of the newly formed Ar–Ar′ bond may be efficiently controlled using a chiral IrCp-complex C4 in combination with a chiral carboxylic acid co-catalyst. Importantly this reaction operates at temperatures as low as 15 °C, thus warranting the atropostability of the newly accessed products. The expected phosphine-naphthol derivatives 18 were thus isolated in generally high enantioselectivities while the control of the diastereoselectivity, strongly depending on the substitution of the phosphine-oxides 16, is more challenging.
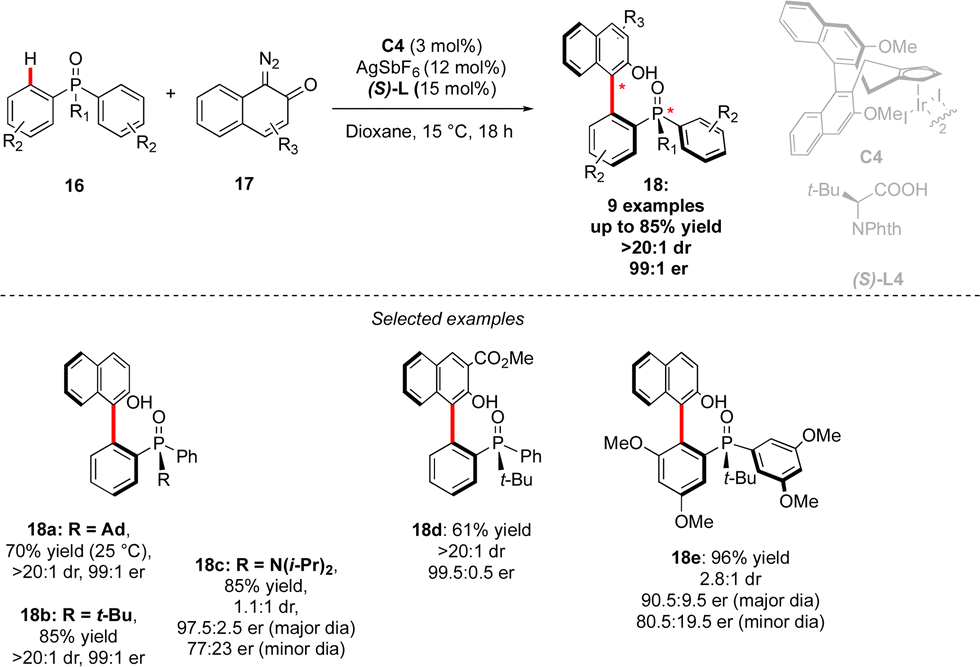 | ||
| Scheme 7 C–C axial chirality + P-point chirality via desymmetrization of phosphine oxide. | ||
An alternative approach to assemble P-chiral/atropisomeric molecules was reported three years later. Similar to the previous example, desymmetrizative C–H functionalization of pro-chiral P(V)-substrates operates, followed by coupling with alkynes and a Satoh–Miura-type annulation step,19 leading to the construction of a sterically hindered naphthyl motif (Scheme 8).20 The afforded compounds 21 feature thus both expected chirality elements. The use of chiral Rh-catalysts warrants efficient chirality transfer, generating 21 with excellent enantioselectivities. Despite the higher reaction temperature (60 °C), increased steric hindrance around the Ar–Ar′ bond (presence of the Ph substituent) translates into augmented atropostability of the newly generated products and thus perfect diastereoselectivities were reached. The mechanistic studies suggest a largely irreversible nature of the C–H cleavage step of the phosphinamide substrate 19 (intermediate A). This first C–H activation is therefore believed to be a turnover-limiting and enantio-determining step with respect to the P-stereogenic center. Insertion of the first alkyne coupling partner into the Rh–C bond (C), followed by cis–trans isomerization delivers the Rh(III) vinyl-intermediate. A second C–H activation thus occurs rapidly, generating atropostable Rh-intermediate D. The insertion of the second diphenylacetylene molecule occurs with the retention of the axial chirality, and reductive elimination delivers the desired product with two perfectly controlled chirality elements.
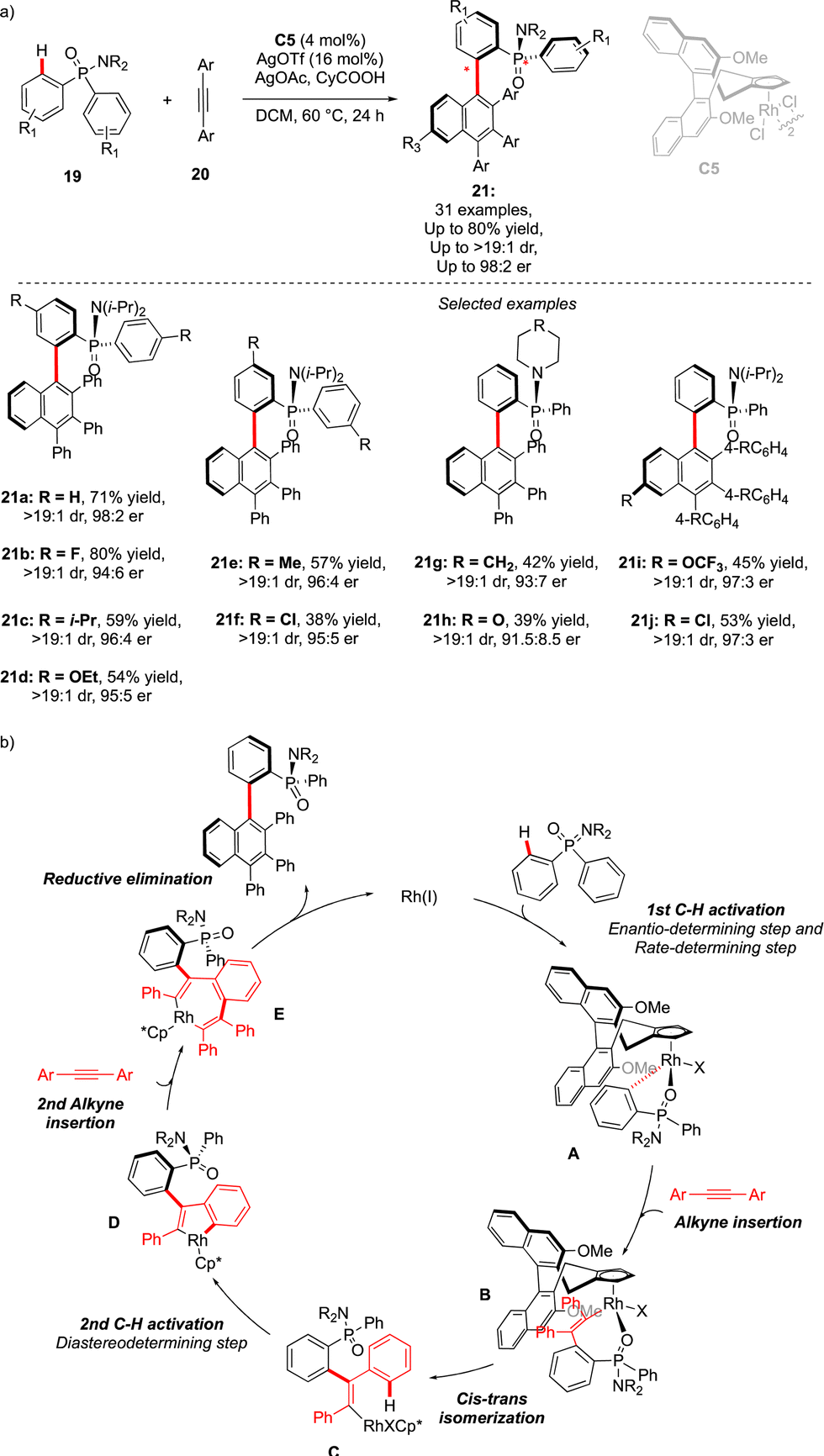 | ||
| Scheme 8 Chiral CpXRh-catalyzed reaction delivering scaffolds featuring C–C atropoisomerism + P-point chirality via construction of the aromatic motif. | ||
A similar approach was also used by Duan to assemble P- and axially chiral phosphine oxides 23 (Scheme 9).21 In this case, desymmetrization of prochiral diaryl-alkyl phosphine oxides 22 could be achieved using a combination of an achiral Cp*Ir-complex and a chiral amide type ligand L5, followed by efficient coupling with alkyne in a Satoh–Miura-type annulation step, furnishing biaryl phosphine oxides in good enantioselectivities and diastereoselectivities and yields up to 92%.
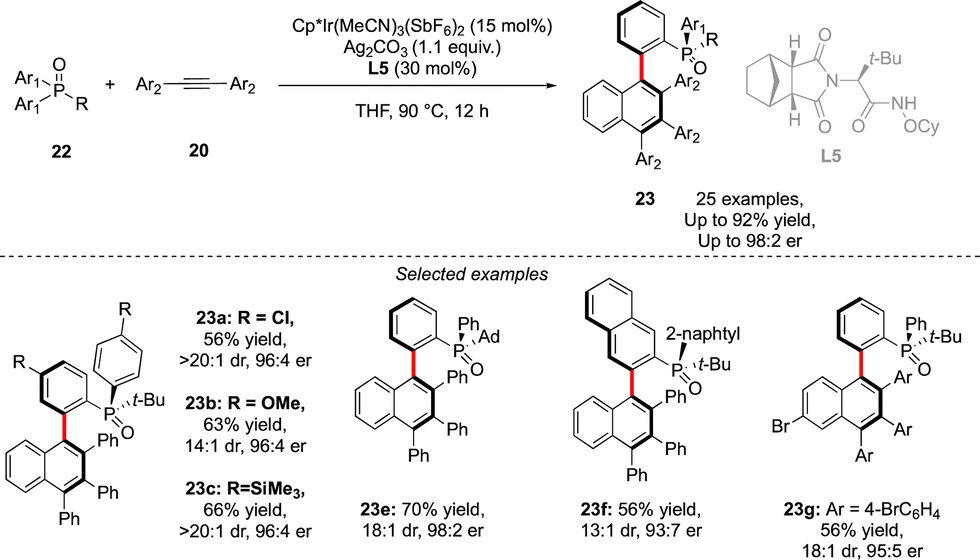 | ||
| Scheme 9 Achiral Cp*Ir-complex + chiral amide ligand catalytic system to assemble scaffolds featuring C–C atropoisomerism + P-point chirality via construction of the aromatic motif. | ||
C–H activation strategy for one-step synthesis of molecules bearing two chiral axes
Owing to the strong dependence of the axial chirality on the reaction temperature and scarcity of the atroposelective direct arylation protocols,22 simultaneous control of two chiral axes during one C–H activation reaction appears as an extremely daunting target. Interesting examples of such transformations were reported by Cramer and Baudoin,22b and Shi.23 In both cases, however, the C–H arylation substrates were designed to allow double independent direct-arylation reactions, i.e. coupling at two distinct positions, thus furnishing final molecules featuring two chiral axes (Scheme 10). However, generation of the double atropoisomerism in a single transformation is infinitely more problematic.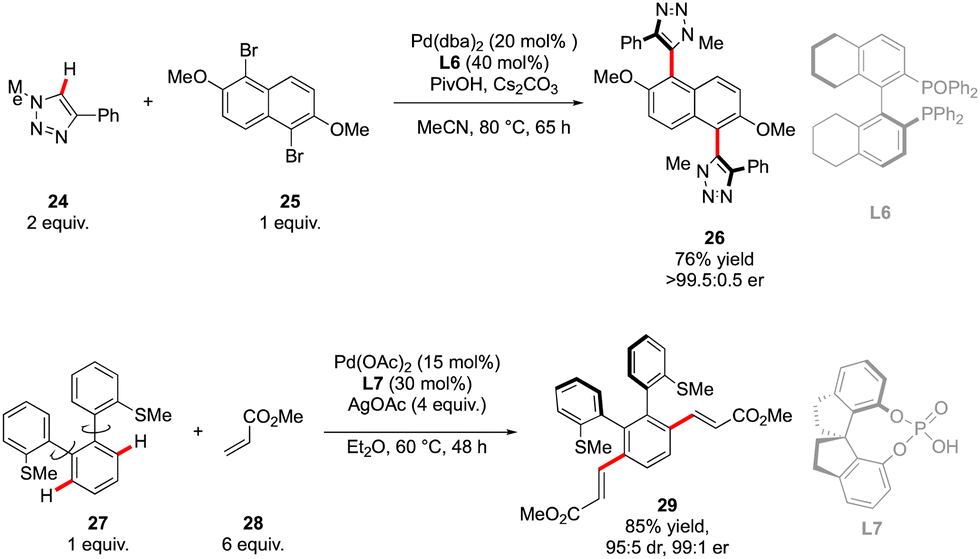 | ||
| Scheme 10 Examples of reaction delivering two chiral elements but occurring as two independent transformations. | ||
The first example of such a transformation was reported by Wencel-Delord and Colobert in 2018.24 The strategy, based on the use of sulfoxide as an enantiopure chiral auxiliary and directing group, implies the functionalization of existing biaryl-sulfoxide precursors.25 However, contrary to the examples described previously (Scheme 2) when alkylation-type reactions were operating to restrict the Ar–Ar′ rotation and deliver central-chirality on the prochiral olefin moiety, the authors focused their attention on developing a direct arylation protocol. Accordingly, Pd-catalyzed coupling between biarylsulfoxide 30 precursors and ortho-substituted aryl iodides 31 was investigated (Scheme 11a). Extensive optimizations of the reaction conditions revealed that the desired arylation may be attainted under the addition of an achiral NHC-type ligand and using two different silver salts. While using the optimized protocol, the desired reactivity could be demonstrated and an almost exclusive formation of a single stereomer out of four possible was observed. A library of the expected enantiomerically pure ortho-triaryl scaffolds was prepared in synthetically useful yields, thus furnishing the original stereogenic scaffolds. From the mechanistic viewpoint, the chirality of the first biaryl-axis is induced during the atroposelective metallation event, by minimizing the steric interactions between the pTol-substituent of the sulfoxide directing group and the NHC-coordinated Pd-catalyst (Scheme 11b). The configuration of the newly generated Ar–Ar′ bond is controlled probably in the next step, i.e. oxidative addition of 31 or a reductive elimination event. The synthetic potential of this transformation was subsequently demonstrated, focusing on the product 32c. The presence of Br- and SOpTol-substituents at the strategic positions gives a unique opportunity to directly convert these scaffolds into topologically unprecedented diphosphine ligands BiaxPhos.
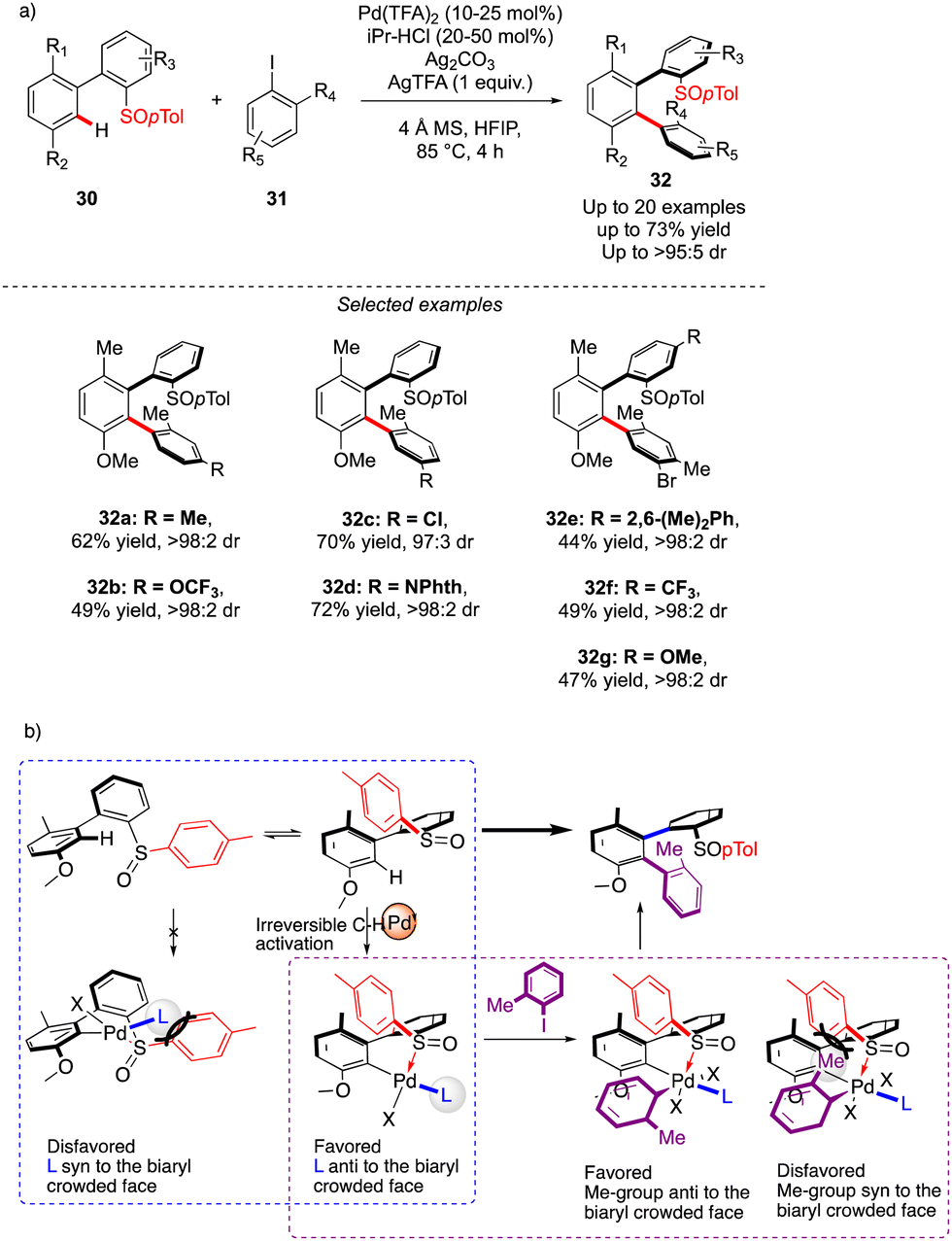 | ||
| Scheme 11 Sulfoxide-directed atroposelective synthesis of triphenyl compounds with two perfectly controlled chiral axes. | ||
While in this seminal work, one chiral axis was induced via functionalization of an already existing biaryl precursor and the second one was assembled via atroposelective direct Ar–Ar′ coupling, synthesis of related triaryl molecules via two Ar–Ar bond formation events is particularly challenging. Zhou proposed an elegant solution to solve this synthetic issue by wisely implementing a Catellani-type approach (Scheme 12).26 Following the principle of Catellani reaction,27 the presence of a norbornene-type ligand should enhance the two-step sequential arylations, ideally with an efficient chirality control. Following such hypothesis, iodoarene 33 was reacted with methyl 2-bromo-3-methylbenzoate and naphthyl trifluoroborates, in the presence of Pd-precatalyst and a chiral norbornene L8. Remarkably, the desired double arylation works beautifully, furnishing the expected vicinal triaryl scaffolds in high yields and excellent diastereo- and enantioselectivities. As expected, the key steps of the catalytic cycle include first Pd(0)-catalyzed direct arylation, followed by norbornene insertion and Pd-migration resulting in the second atroposelective Ar–X/Ar–H coupling. The high level of chirality transfer from the norbornene and the operational simplicity of the overall transformation is truly impressive.
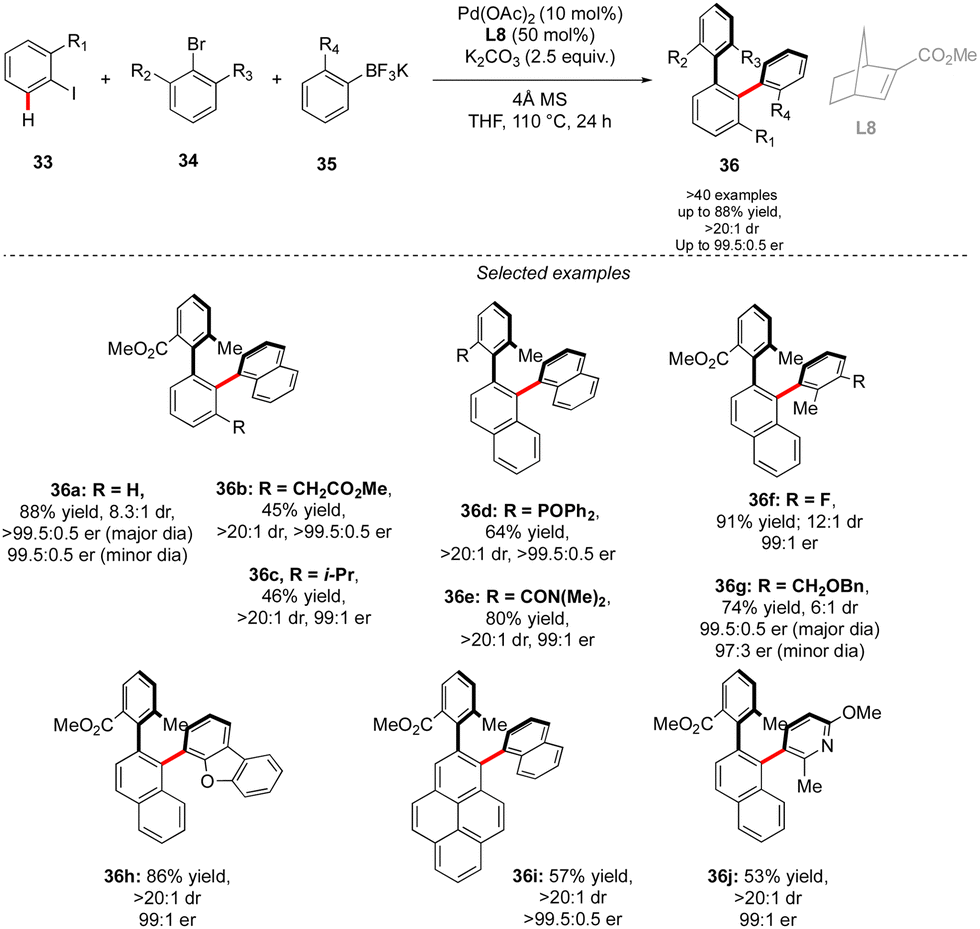 | ||
| Scheme 12 Synthesis of triaryl compounds via double atroposelective direct arylation. | ||
A complementary strategy to build up vicinal triaryl motifs with perfect control of both chiral axes was recently demonstrated by Hong, Yao and Shi (Scheme 13).28 In this case an intramolecular reaction was designed, implying a C–H annulation-type protocol. The very precise design of substrate 37 is the cornerstone of this approach. The quinoline motif used as the directing group furnishes one Ar-motif while the second aromatic ring is installed at the terminal position of the ether-linked alkyne. Accordingly, amido-quinoline-directed C–H activation using Co-precatalyst, Salox-type ligand L9 and Mn(OAc)3 takes place, and the intramolecular migratory insertion of the tethered alkyne, followed by the annulation step, furnished 38. The reaction is limited by the substrate design but certain variability may still be introduced by diversifying electronic- and steric-properties of the substituents placed on the two phenyl moieties.
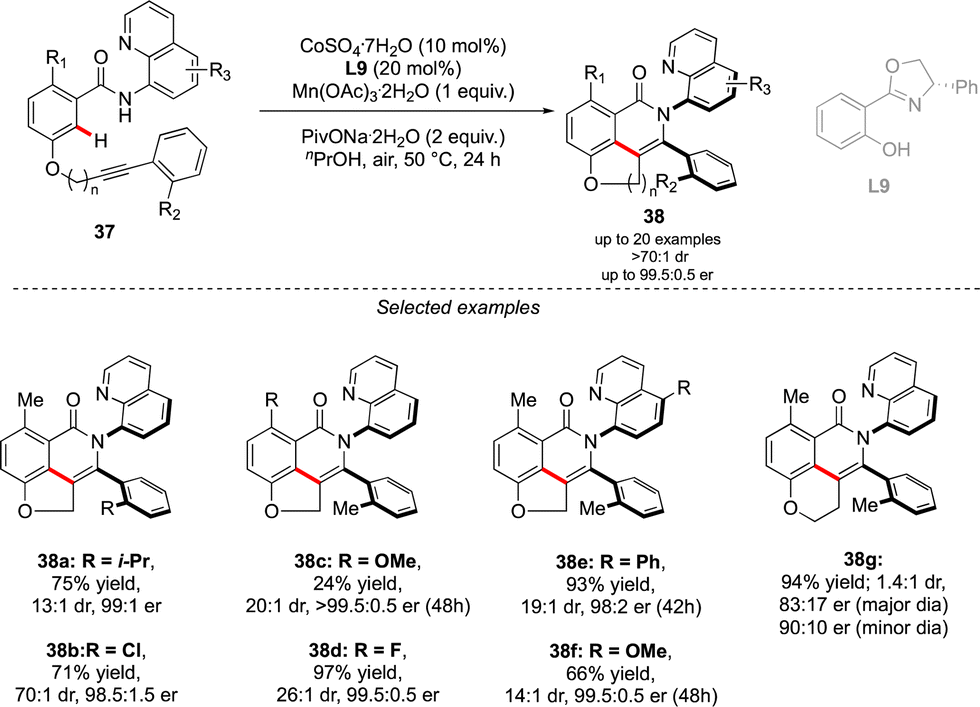 | ||
| Scheme 13 Synthesis of triphenyl with vicinal atropisomeric axes via intramolecular coupling. | ||
Axial chirality and planar chirality via C–H activation
Very recently, the concept of the multichiral atropisomeric molecules has been extended even further, by combining both the axial and planar chiralities. To assemble such compounds via a C–H activation strategy, the group of Cheng and Zhou drew inspiration from the double-atroposelective reactions performed using the Catellani type approach. It was hypothesized that a precisely designed aryl-ferrocene-type substrate would react smoothly with ortho-,ortho′-substituted aryl bromide to undergo first atroposelective Ar–Ar bond formation, with chiral information dictated by the presence of a chiral norbornene ligand (Scheme 14).29 The atropisomeric character of the biaryl would further induce, via an unprecedented axial-to-planar diastereoinduction process, the planar chirality, thus delivering the desired multichiral molecules. This working hypothesis was rapidly validating, while reacting ferrocene derived 39 with 34. Remarkably, combination of an achiral phosphine MePhos ligand and enantiopure norbornene L10 was essential to promote the desired pathway, while avoiding possible side reactions such as premature direct cyclization of 39 or direct arylation of the ferrocene substrate. Under the optimized reaction conditions, this catalytic system showed exceptional efficiency, furnishing a large panel of five- to seven-membered benzo-fused ferrocenes with both axial and planar chiralities with constantly excellent enantioselectivity of 99% and very high diastereoselectivity. The reaction tolerated not only a diversity of aryl-bromides as the coupling partners, but also both ferrocene- and ruthenacene scaffolds.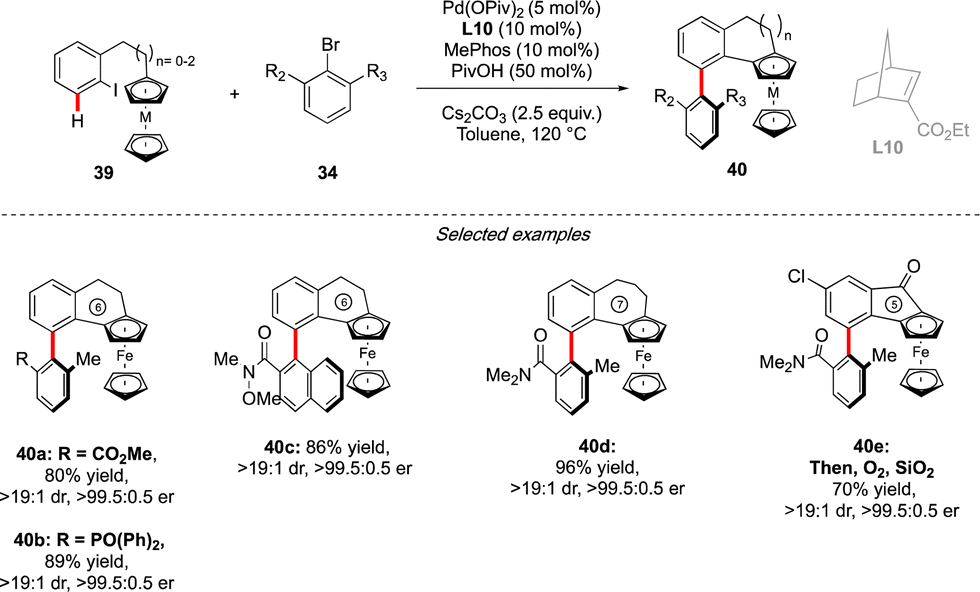 | ||
| Scheme 14 Axial + planar chirality via Catellani approach. | ||
Conclusions
With respect to the continuously expanding need for chiral scaffolds, the synthesis of topologically unique and complex molecules will certainly continue to be a rapidly evolving field of synthetic chemistry. The C–H activation approach, allowing direct incorporation of functional groups or molecular motifs at specific positions, gives therefore unique opportunities to assemble enantiopure molecules, bearing even two chiral elements, within one stereoselective transformation. The recent examples, detailed in this review article, describe the possibilities offered by the C–H activation mindset to rapidly afford a diversity of compounds bearing either two chiral axes or one chiral axis and one point chirality. Interestingly, the large structural diversity of such compounds results from mechanistically different approaches envisioned over the last few years. Accordingly, within the near future, alternative catalytic systems will certainly be designed to further expand this emerging field. Targeting even more sustainable transformations and benefiting from the rising knowledge on asymmetric 3d-metal catalyzed asymmetric C–H activation, alternative multi-stereoselective transformations catalyzed by more abundant and less toxic metals, including Co, Fe, Ni, or Cu, for example, will certainly catch the attention of the scientific community. In parallel, the synthesis of topologically alternative structures, combining other chirality elements such as planar chirality, atropoisomerism arising from the restricted rotation around the Ar-olefin bond and various point chiralities is to be expected, thus furnishing appealing chiral products for biological and/or material science applications.Author contributions
Amandine Luc participated in the conception of the article and writing of the first draft. Joanna Wencel-Delord was involved in the conception of the article, writing, and correcting the final version of the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. W.-D. thanks the CNRS (Centre National de la Recherche Scientifique), the “Ministere de l’Education Nationale et de la Recherche“, France, for financial support. A. L. and J. W.-D. acknowledge the European Commission for the ERC-Starting Grant “AlCHIMIE” no. 949804.Notes and references
- J. Ceramella, D. Iacopetta, A. Franchini, M. De Luca, C. Saturnino, I. Andreu, M. S. Sinicropi and A. Catalano, Appl. Sci., 2022, 12, 10909 CrossRef CAS.
- (a) J. R. Brandt, F. Salerno and M. J. Fuchter, Nat. Rev. Chem., 2017, 1, 0045 CrossRef CAS; (b) B. Zhao, S. Yang, J. Deng and K. Pan, Adv. Sci., 2021, 8, 2003681 CrossRef CAS PubMed; (c) P. Jeschke, Pest. Manag. Sci., 2018, 74, 2389–2404 CrossRef CAS PubMed.
- X. Bao, J. Rodriguez and D. Bonne, Angew. Chem., Int. Ed., 2020, 59, 12623–12634 CrossRef CAS PubMed.
- G. Beutner, R. Carrasquillo, P. Geng, Y. Hsiao, E. C. Huang, J. Janey, K. Katipally, S. Kolotuchin, T. La Porte, A. Lee, P. Lobben, F. Lora-Gonzalez, B. Mack, B. Mudryk, Y. Qiu, X. Qian, A. Ramirez, T. M. Razler, T. Rosner, Z. Shi, E. Simmons, J. Stevens, J. Wang, C. Wei, S. R. Wisniewski and Y. Zhu, Org. Lett., 2018, 20, 3736–3740 CrossRef CAS PubMed.
- For general reviewws on C–H activation see: (a) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC; (b) L. Zhang and T. Ritter, J. Am. Chem. Soc., 2022, 144, 2399–2414 CrossRef CAS PubMed; (c) L. Zhang and T. Ritter, J. Am. Chem. Soc., 2022, 144, 2399–2414 CrossRef CAS PubMed.
- For general reviews see: (a) D. J. Abrams, P. A. Provencher and E. J. Sorensen, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC; (b) N. Y. S. Lam, K. Wu and J. Yu, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202011901; (c) J. Börgel and T. Ritter, Chem, 2020, 6, 1877–1887 CrossRef; (d) D. Basu, S. Kumar, S. Sudhir V and R. Bandichhor, J. Chem. Sci., 2018, 130, 71 CrossRef.
- For selected reviews see: (a) F. Colobert and J. Wencel-Delord, C–H activation for asymmetric synthesis, 2019 CrossRef; (b) C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS PubMed; (c) J. Wencel-Delord and F. Colobert, Synlett, 2015, 2644–2658 CrossRef CAS; (d) Ł. Woźniak and N. Cramer, Trends Chem., 2019, 1, 471–484 CrossRef; (e) J. Loup, U. Dhawa, F. Pesciaioli, J. Wencel-Delord and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 12803–12818 CrossRef CAS PubMed; (f) G. Liao, T. Zhang, Z. Lin and B. Shi, Angew. Chem., Int. Ed., 2020, 59, 19773–19786 CrossRef CAS PubMed.
- F. Kakiuchi, P. Le Gendre, A. Yamada, H. Ohtaki and S. Murai, Tetrahedron: Asymmetry, 2000, 11, 2647–2651 CrossRef CAS.
- (a) J. Wencel-Delord and F. Colobert, SynOpen, 2020, 04, 107–115 CrossRef CAS; (b) G. Liao, T. Zhou, Q.-J. Yao and B.-F. Shi, Chem. Commun., 2019, 55, 8514–8523 RSC; (c) T. K. Achar, S. Maiti, S. Jana and D. Maiti, ACS Catal., 2020, 10, 13748–13793 CrossRef CAS; (d) C.-X. Liu, W.-W. Zhang, S.-Y. Yin, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2021, 143, 14025–14040 CrossRef CAS PubMed; (e) J. A. Carmona, C. Rodríguez-Franco, R. Fernández, V. Hornillos and J. M. Lassaletta, Chem. Soc. Rev., 2021, 50, 2968–2983 RSC.
- For selected examples see: (a) J. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2014, 53, 13244–13247 CrossRef CAS PubMed; (b) Q. Wang, W.-W. Zhang, H. Song, J. Wang, C. Zheng, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2020, 142, 15678–15685 CrossRef CAS PubMed; (c) Q. Wang, Z.-J. Cai, C.-X. Liu, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2019, 141, 9504–9510 CrossRef CAS PubMed; (d) J. Zheng, W.-J. Cui, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2016, 138, 5242–5245 CrossRef CAS PubMed.
- A. Romero-Arenas, V. Hornillos, J. Iglesias-Sigüenza, R. Fernández, J. López-Serrano, A. Ros and J. M. Lassaletta, J. Am. Chem. Soc., 2020, 142, 2628–2639 CrossRef CAS PubMed.
- D. Liang, J.-R. Chen, L.-P. Tan, Z.-W. He and W.-J. Xiao, J. Am. Chem. Soc., 2022, 144, 6040–6049 CrossRef CAS PubMed.
- For an additional example of organocatalyzed transformation affording multichiral molecule bearing Ar–Ar axis and central-chirality see: H.-Q. Wang, S.-F. Wu, J.-R. Yang, Y.-C. Zhang and F. Shi, J. Org. Chem., 2022 DOI:10.1021/acs.joc.2c02303.
- Y. Li, Y. Liou, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2022, 61, e202212595 CAS.
- J. Wang, H. Chen, L. Kong, F. Wang, Y. Lan and X. Li, ACS Catal., 2021, 11, 9151–9158 CrossRef CAS.
- F. Wang, J. Jing, Y. Zhao, X. Zhu, X. Zhang, L. Zhao, P. Hu, W. Deng and X. Li, Angew. Chem., Int. Ed., 2021, 60, 16628–16633 CrossRef CAS PubMed.
- J. Diesel and N. Cramer, ACS Catal., 2019, 9, 9164–9177 CrossRef CAS.
- Y.-S. Jang, Ł. Woźniak, J. Pedroni and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 12901–12905 CrossRef CAS PubMed.
- N. Umeda, H. Tsurugi, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2008, 47, 4019–4022 CrossRef CAS PubMed.
- W. Xia, Q. An, S. Xiang, S. Li, Y. Wang and B. Tan, Angew. Chem., Int. Ed., 2020, 59, 6775–6779 CrossRef CAS PubMed.
- C.-W. Zhang, X.-Q. Hu, Y.-H. Dai, P. Yin, C. Wang and W.-L. Duan, ACS Catal., 2022, 12, 193–199 CrossRef CAS.
- (a) K. Yamaguchi, H. Kondo, J. Yamaguchi and K. Itami, Chem. Sci., 2013, 4, 3753–3757 RSC; (b) Q.-H. Nguyen, S.-M. Guo, T. Royal, O. Baudoin and N. Cramer, J. Am. Chem. Soc., 2020, 142, 2161–2167 CrossRef CAS PubMed; (c) C. G. Newton, E. Braconi, J. Kuziola, M. D. Wodrich and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 11040–11044 CrossRef CAS PubMed; (d) Ł. Woźniak and N. Cramer, Angew. Chem., Int. Ed., 2021, 60, 18532–18536 CrossRef PubMed; (e) N. Jacob, Y. Zaid, J. C. A. Oliveira, L. Ackermann and J. Wencel-Delord, J. Am. Chem. Soc., 2022, 144, 798–806 CrossRef CAS PubMed.
- (a) G. Liao, T. Zhang, L. Jin, B. Wang, C. Xu, Y. Lan, Y. Zhao and B. Shi, Angew. Chem., Int. Ed., 2022, 61, e202115221 CAS; (b) L. Jin, P. Zhang, Y. Li, X. Yu and B.-F. Shi, J. Am. Chem. Soc., 2021, 143, 12335–12344 CrossRef CAS PubMed.
- Q. Dherbassy, J.-P. Djukic, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2018, 57, 4668–4672 CrossRef CAS PubMed.
- For other examples of atroposelective C–H activation using biaryl-sulfoxides see: (a) T. Wesch, F. R. Leroux and F. Colobert, Adv. Synth. Catal., 2013, 355, 2139–2144 CrossRef CAS; (b) C. K. Hazra, Q. Dherbassy, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2014, 53, 13871–13875 CrossRef CAS PubMed; (c) Q. Dherbassy, G. Schwertz, M. Chessé, C. K. Hazra, J. Wencel-Delord and F. Colobert, Chem. – Eur. J., 2016, 22, 1735–1743 CrossRef CAS PubMed; (d) Q. Dherbassy, J. Wencel-Delord and F. Colobert, Tetrahedron, 2016, 72, 5238–5245 CrossRef CAS.
- Q. Gao, C. Wu, S. Deng, L. Li, Z.-S. Liu, Y. Hua, J. Ye, C. Liu, H.-G. Cheng, H. Cong, Y. Jiao and Q. Zhou, J. Am. Chem. Soc., 2021, 143, 7253–7260 CrossRef CAS PubMed.
- M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed. Engl., 1997, 36, 119–122 CrossRef CAS.
- B. Wang, G. Xu, Z. Huang, X. Wu, X. Hong, Q. Yao and B. Shi, Angew. Chem., Int. Ed., 2022, 61, e202208912 CrossRef CAS PubMed.
- J. Ye, L. Li, Y. You, C. Jiao, Z. Cui, Y. Zhang, S. Jia, H. Cong, S. Liu, H.-G. Cheng and Q. Zhou, JACS Au, 2023, 3, 384–390 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |