
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-assembly of a fluorescent hydrogen-bonded capsule based on an amino-acid functionalised tetraphenylethylene†
Anna
Brzechwa-Chodzyńska‡
 ab,
Grzegorz
Markiewicz‡
ab,
Piotr
Cecot
ab,
Jack
Harrowfield
c and
Artur R.
Stefankiewicz
*ab
ab,
Grzegorz
Markiewicz‡
ab,
Piotr
Cecot
ab,
Jack
Harrowfield
c and
Artur R.
Stefankiewicz
*ab
aFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznańskiego 8, Poznań 61-614, Poland. E-mail: ars@amu.edu.pl
bCenter for Advanced Technology, Adam Mickiewicz University, Uniwersytetu Poznańskiego 10, Poznań 61-614, Poland
cUniversité de Strasbourg, ISIS, 8 allée Gaspard Monge, Strasbourg 67083, France
First published on 28th April 2023
Abstract
Herein we report the self-assembly of an amino-acid substituted tetraphenylethylene (TPE) into a hydrogen-bonded dimeric capsule. Depending on the conditions applied the presented TPE derivative exhibits bluish photoluminescence with QYs up to 24% due to the AIE effect and excimer formation.
Stimuli-responsive luminogenic materials have attracted considerable attention owing to their numerous prominent photophysical features and a wide range of applications.1–5 Among them, fluorescent three-dimensional molecular and supramolecular capsules have emerged as particularly promising candidates for the sensing materials of tomorrow.6,7 This is due to possible applications in biology, medical and environmental fields that have the potential of developing into real-life technologies.
Over the past decades, substantial progress has been made in the ability to design and assemble fluorescent capsules via covalent (reversible and irreversible) bonds or supramolecular interactions such as coordinative and hydrogen bonds, the last being exemplified in this work.8–13 The vast majority of fluorescent hydrogen-bonded capsules have been constructed from phenol-based macrocycles, namely, resorcinarenes, and/or exist only in the presence of stabilizing template molecules. The generation of fluorescent capsules via formation of hydrogen bonds between non-macrocyclic components is much more challenging and rare.14,15 A means of overcoming this difficulty is to use photoactive building blocks pre-designed to form exclusively dimeric capsular entities, although there is disadvantage to this approach in that large capsules require building blocks that are no longer simple to synthesize. An alternative is to use “chirally restricted” building blocks to limit the possible forms of aggregation.16
Fluorophores that are non-emissive in dilute solution but exhibit strong fluorescence in the aggregated state due to the restriction of molecular rotation of their aryl groups are of especial interest for capsule synthesis.17 Two decades ago, Tang and co-workers termed this effect “aggregation-induced emission” (AIE).18,19 Tetraphenylethene (TPE)20 is a promising building block for the construction of luminescent macrocycles and cages,12,13 due to its simple but stable chemical structure, C2 symmetry, quadruple reaction sites, and strong AIE effect.21 A study20 of different methylated derivatives of TPE has demonstrated the operation of the “restricted intramolecular rotation” (RIR) mechanism in controlling their AIE behavior, the restrictions arising from steric hindrance in the isolated molecules as well as in aggregates formed in solution and the solid state. Factors such as solvent viscosity,22 molecular cyclization,23 host–guest interactions,24 hydrogen bonding.18 and many others25,26 have also been recognized as important influences on aryl group rotation.
Although many molecules based on AIE-gens have been reported in the past decade,19 no fluorescent nanocapsules held together by hydrogen bonds have been reported. Previously, we have shown that addition of amino-acid arms to a 1,3,5-benzene tricarboxylate core can be used to obtain an octameric hydrogen-bonded capsule.25 To create a fluorescent supramolecular capsule, we decided to utilize TPE as an aromatic core and to functionalize it with homochiral amino-acid substituents which, in addition to providing sites for H-bonding interactions, should induce “chiral restrictions” on the outcoming aggregate. Thus, we describe here a newly designed hydrogen bonded chiral nanocapsule, formed by the self-assembly of an amino-acid functionalized TPE in a weakly polar solvent. The supramolecular capsule is held together by four hydrogen bonding pairs between two enantiomerically pure components that consist of an aromatic TPE unit equipped with four amino-acid (S-trityl-L-cysteine) functional arms acting as both H-bond acceptors and donors (Fig. 1). The nanocapsule features chiral luminescence and simultaneous chiroptical characterization based on fluorescence detected circular dichroism (FDCD) and electronic circular dichroism (ECD) has been performed for determination of the thermodynamic parameters governing its self-assembly. This TPE derivative exhibits a remarkable AIE effect, with up to 16-times increase in photoluminescence quantum yield in its aggregated state.
 | ||
| Fig. 1 Chemical structures (top) and schematic representations (bottom) of the self-assembly of 1 into dimeric nanocapsule 12 in TCE. | ||
In extension of our previous work where the trityl protecting group on L-cysteine appeared beneficial in enhancing both the solubility and stability of an octameric capsule in apolar solvents,25 TPE-S-trityl-L-cysteine (1) was synthesized by connection of the TPE aromatic core with four terminally located S-trityl-L-cysteine units (see ESI† for details). Confirmation of the expected composition of 1 was provided by NMR spectroscopy as well as mass spectrometry and elemental analysis (Fig. S3–S7 in ESI†).
1 was found to be soluble in highly polar solvents like DMSO or THF, while poorly soluble in most solvents of low polarity (e.g. chloroform, dichloromethane, benzene or toluene), which are non-competitive media for the formation of inter- (or intra-) molecular hydrogen bonds. Ultimately, 1,1,2,2-tetrachloroethane (TCE) was employed to study the self-assembly process of 1 in solution, as this solvent does not disrupt hydrogen bonding interactions and has wide liquid range (−44 to 146 °C) making it particularly suitable for variable temperature measurements.
The formation of a supramolecular capsule 12 in solution was firstly detected by 1H NMR spectroscopy. The 1H NMR spectra were recorded in three solvents (all at C = 2.0 × 10−3 M) differing markedly in polarity and hydrogen bond formation capacity i.e. polar: THF-d8 (Fig. 2a) and DMSO-d6 (Fig. S3 in ESI†); and apolar TCE-d2 (Fig. 2b), the latter, as noted above, being a poor hydrogen bond acceptor but one in which 1 showed sufficient solubility. The recorded spectra differed significantly from each other, the strong broadening of the peaks in TCE-d2 contrasting with the relatively sharp spectra in THF-d8 and DMSO-d6. These results parallel other studies conducted by e.g. Takeda and co-workers where benzeneoligoamides27 and pyrenetetraamide28 were found to exhibit broad signals of protons due to hydrogen bonding driven aggregation. Apart from broadening effect, upon formation of the hydrogen bonded capsule 12, indicative changes were also observed for the amide-NH (marked c) and α-CH (marked d) signals, which shift upfield by 1.3 ppm and 0.2 ppm, respectively, while the outer TPE resonance (marked b) shifts downfield by 0.3 ppm. Significantly, the upfield shift of the amide-NH signal indicates that the amido groups are not involved in the self-assembly process, as downfield shifts only were observed in highly polar solvents (i.e. THF-d8, DMSO-d6) where those protons would be involved in solvation interactions. The apparent splitting of this resonance in THF-d8 might be attributed to incomplete solvation of the NH sites, as this effect was not observed in the more strongly solvating DMSO-d6 (Fig. S3 in ESI†) while the diffusion ordered spectrum (DOSY-NMR) recorded in THF-d8 showed all signals originating from the same species (Fig. S12 in ESI†).
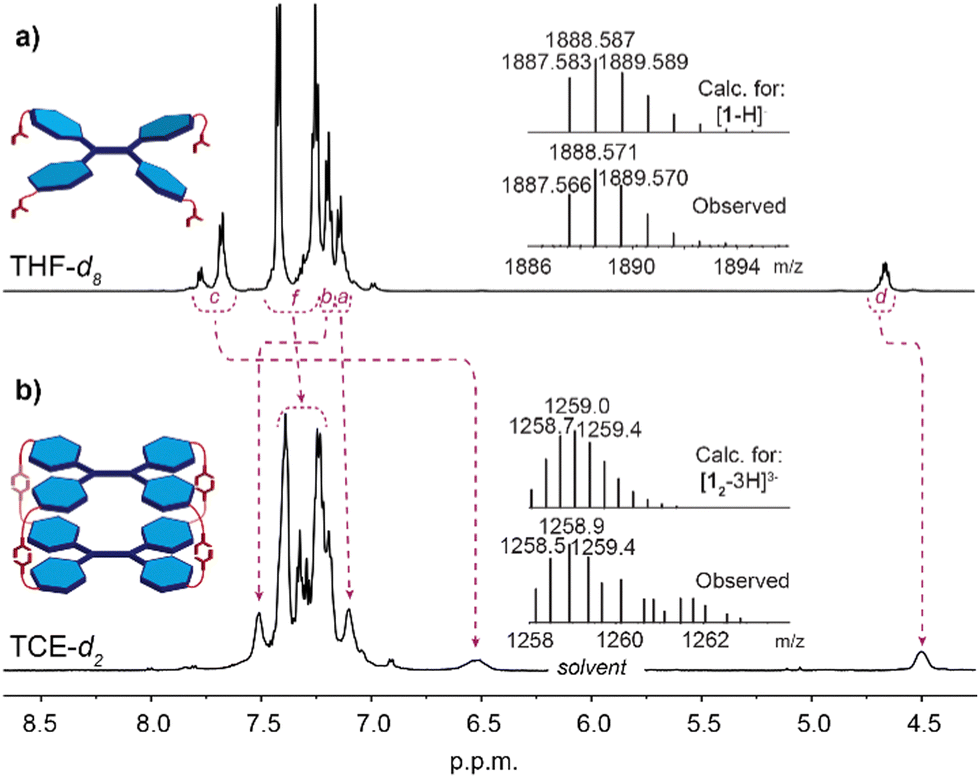 | ||
| Fig. 2 1H NMR (600 MHz) spectra at 298 K of 1 (C = 2.0 × 10−3 M). Recorded in: (a) THF-d8. Inset: ESI-TOF-MS spectrum of 1; (b) TCE-d2. Inset: MALDI-MS spectrum of 12. | ||
All attempts to confirm the composition of the capsule in the gas phase by ESI-mass spectrometry were unsuccessful, which was attributed to the disassembly of the non-covalent capsule 12 under the experimental conditions employed. Ions corresponding to 12 were observed by MALDI mass spectrometry operating in linear mode, which showed a negatively charged isotopic clusters centred at m/z = 1259 for [12-3H]3−, m/z = 1890 for [12-2H]2− and m/z = 3776 for [12-H]−, the latter one being weak due to unfavourable ionization pathway under MALDI conditions (Fig. 2b, and Fig. S8 in ESI†).
That the self-assembly reaction was that of a dimeric architecture was further confirmed by DOSY NMR. Again, spectra in TCE-d2 (Fig. 3) and THF-d8 (Fig. S12 in ESI†) were very different, although both showed evidence of the presence of a single species. Using the Stokes–Einstein equation, the solvodynamic radius of the species present in TCE-d2 was calculated to be 13.7 Å (D = 1.1 × 10−10 m2 s−1), which is in excellent agreement with the predicted value for the dimeric capsule 12 based on its calculated structure (Fig. 3 inset). In contrast, solvodynamic radius recorded in THF-d8 was significantly smaller and equal to 6.3 Å (D = 7.53 × 10−10 m2 s−1) corresponding well with the value expected for the single component of 1.
 | ||
| Fig. 3 DOSY NMR (600 MHz, TCE-d2) spectrum of 1 recorded at C = 2.0 × 10−3 M and 298 K. Solvodynamic radius calculated according to the Stockes–Einstein equation. Inset: Calculated (semi-empirical) structure of 12. | ||
Based on the methodology utilized by Mastalerz and co-workers16 for H-bonded nanocapsules, semi-empirical RM1 modelling was performed for our system (Fig. 3, inset). As a chiral species, the dimeric capsule was expected to be of D2 symmetry. The anticipated structure allowed for the involvement of both NH⋯O and OH⋯O bonds between amino acid side chains but the RM1 modelling revealed only four OH⋯O hydrogen bonding pairs between COOH units, leaving all CONH amide units unused due to structural constraints. The hydrogen bonds had a typical length for an OH⋯O motif of ∼2.6 Å between the two oxygen atoms.29 The calculated array of hydrogen bonds found its full confirmation in the IR spectrum, where an NH stretch band for unbound amide groups was observed at 3450 cm−1, while a broad and shifted OH band appeared at 2600 cm−1 (Fig. S18 in ESI†).26,30
The inherent chirality of 1 means that measurements of rotational strength can be applied to study of its aggregation behaviour. In particular, the circular dichroism (CD) spectrum of 1 in TCE shows a very marked temperature dependence, indicating that it could be used as a sensitive quantitative measure of the monomer–dimer equilibrium (Fig. 4a). The changes in ellipticity at λ = 300 nm as a function of temperature were sigmoidal, a form characteristic of either simple dimerization or an equal-K isodesmic self-assembly process (complete data shown in Fig. 4b). Although both processes are indistinguishable from the fitting, the apparent DOSY size leaves dimerization as the only possible outcome of the assembly. The dimerization of 1 is clearly enthalpy driven, as indicated by the positive slope of the corresponding van’t Hoff plot (Fig. S19 in ESI†), with the calculated enthalpy change of ΔH = −65 ± 0.4 kJ mol−1, entropy change of ΔS = −140 ± 1.4 J K−1 mol−1 and the dimerization equilibrium constant of K298 K = 1.2 × 104 M−1 (Fig. 4b).
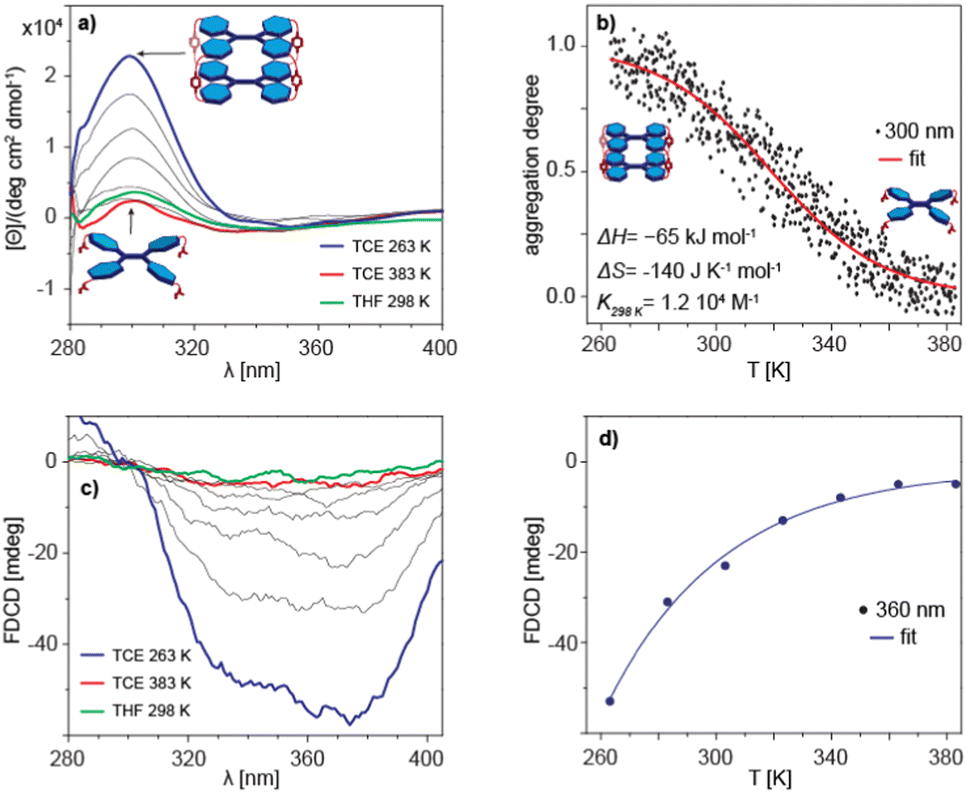 | ||
| Fig. 4 (a) VT CD spectra of 1 recorded in TCE and THF at C = 5.0 × 10−4 M (b) VT-CD intensity cooling scans followed at λ = 300 nm (cooling rate= −1 K min−1). (c) VT FDCD spectra of 1 recorded in TCE and THF at C = 1.0 × 10−4 M, LP filter = 420 nm, HT = 700 V. (d) VT FDCD intensity cooling scans followed at λ = 360 nm (cooling rate = −1 K min−1). | ||
Similar analysis performed on the temperature dependence of the fluorescence-detected circular dichroism (FD CD) gave rather unexpected results (Fig. 4c). The change in intensity upon cooling did not have the sigmoidal form anticipated for a dimerization process (Fig. 4d). The lack of a correlation between aggregation and emission indicates that the species actually exhibiting photoluminescence are not the monomers or dimers themselves, but rather that 1 upon excitation forms different entities, e.g. a fluorescent excimer, similar to that discussed by Takeda et al.31
In order to verify this, concentration-dependent photoluminescence spectra of 1 in TCE were recorded (Fig. 5a). 1 when dissolved in TCE exhibits bright bluish fluorescence upon UV excitation (λex = 390 nm), with two overlapping emission bands at λem = 480 nm and λem = 520 nm. Surprisingly, the photoluminescence intensity was found to be linearly dependent on the concentration up to C = 1.0 × 10−4 M above which the PL spectra are disrupted by internal filter effects (Fig. 5b). The emission quantum yield of QY = 6.1% was determined by a comparative method, using quinine sulfate (QS) as a standard.
 | ||
Fig. 5 (a) Concentration dependent UV-vis (black) and PL (blue, λex = 390 nm) spectra of 1 recorded in TCE at 298 K. (b) Determination of a photoluminescence QY of 1 in TCE. Inset: Photography of the solution of 1 in TCE upon UV light irradiation. (c) PL (blue, λex = 390 nm) spectra of 1 recorded in THF/H2O mixtures (C = 1.0 × 10−5 M). Inset: UV-vis (black) spectrum of 1 in THF. (d) Changes in the photoluminescence QY of 1 upon increase in the concentration of H2O in THF. Inset: Photography of the solutions of 1 in THF and H2O/THF (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) upon UV light irradiation. :1) upon UV light irradiation. | ||
Taking into account the results above, one can conclude that the dimerization process in the ground state does not affect the emissive properties of 1 in TCE solution. Instead, the observed emission reflects the excited-state equilibrium involving the formation of an excimer which not only significantly reduces the apparent emission QY down to 6.1%, but also results in an unusual double band emission of 1.
With an unsuccessful attempt to induce AIE-effects by dimerization, we decided to check whether the emission of 1 might be altered by formation of higher aggregates. In contrast to TCE, 1 when dissolved in THF does not exhibit any major photoluminescence (QY = 1.5%) (Fig. 5c and d). Addition of water however can induce strong hydrophobic effects and push 1 towards aggregation. Indeed, upon addition of water a sharp increase in the photoluminescence intensity was observed, with the QY reaching up to 24% indicating the remarkable AIE tendency of 1 (Fig. 5d).
The formation of large aggregates of 1 upon varying the solvent composition was further confirmed by Dynamic Light Scattering (DLS). DLS analysis of 1 recorded in H2O/THF (9:1) revealed the formation of uniform nanoparticles with a diameter of 102 ± 21 nm at C = 1.0 × 10−5 M and 147 ± 40 nm at C = 1.0 × 10−4 M, with polydispersity PI < 0.1 in both cases (Fig. S14 and S15 in ESI†). Further increase in the concentration resulted in precipitation of 1 in the form of an off-white amorphous solid.
In conclusion, we present a newly designed TPE derivative, which due to its functionalization with amino-acid side arms, can self-assemble into a chiral, non-covalent dimeric capsule in solvents of low polarity. The capsule is held together by four pairs of hydrogen bonds formed between amino acid COOH units, leaving amide linkers unoccupied. Surprisingly, the dimerization in the ground state does not affect the fluorescent properties of TPE core, as those are governed by the excited state equilibrium involving excimer species. However, due to the strong AIE tendency of 1, photoluminescence quantum yields up to 24% can be attained in the aggregated state.
A. R. S. thanks to the National Science Centre (grant SONATA BIS 2018/30/E/ST5/00032) for financial support. The work was supported by grant no. POWR.03.02.00-00-I026/16. The calculations were carried out at Poznań Supercomputing and Networking Center, grant no. 401. A. R. S and A. B.-C. would like to thank Prof. Pablo Ballester and his group for their help and knowledge.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Gao and B. Z. Tang, ACS Sens., 2017, 2, 1382–1399 CrossRef CAS PubMed.
- J. Sun, H. Li, X. Gu and B. Z. Tang, Adv. Healthcare Mater., 2021, 10, 2101177 CrossRef CAS PubMed.
- H. Wang, E. Zhao, J. W. Y. Lam and B. Z. Tang, Mater. Today, 2015, 18, 365–377 CrossRef CAS.
- S. Xu, Y. Duan and B. Liu, Adv. Mater., 2020, 32, 1903530 CrossRef CAS PubMed.
- D. Zhang, T. K. Ronson, Y.-Q. Zou and J. R. Nitschke, Nat. Rev. Chem., 2021, 5, 168–182 CrossRef CAS PubMed.
- A. Brzechwa-Chodzyńska, W. Drożdż, J. Harrowfield and A. R. Stefankiewicz, Coord. Chem. Rev., 2021, 434, 213820 CrossRef.
- Ł. Szyszka, M. Górecki, P. Cmoch and S. Jarosz, J. Org. Chem., 2021, 86, 5129–5141 CrossRef PubMed.
- Y. Jiao, Y. Zuo, H. Yang, X. Gao and C. Duan, Coord. Chem. Rev., 2021, 430, 213648 CrossRef CAS.
- J. Zhao, Z. Zhou, G. Li, P. J. Stang and X. Yan, Natl. Sci. Rev., 2021, 8, nwab045 CrossRef CAS PubMed.
- Y. Li, J. Zhang, H. Li, Y. Fan, T. He, H. Qiu and S. Yin, Adv. Opt. Mater., 2020, 8, 1902190 CrossRef CAS.
- X. Jing, C. He, L. Zhao and C. Duan, Acc. Chem. Res., 2019, 52, 100–109 CrossRef CAS PubMed.
- J. Jiao, Z. Li, Z. Qiao, X. Li, Y. Liu, J. Dong, J. Jiang and Y. Cui, Nat. Commun., 2018, 9, 4423 CrossRef PubMed.
- W. Zheng, G. Yang, S.-T. Jiang, N. Shao, G.-Q. Yin, L. Xu, X. Li, G. Chen and H.-B. Yang, Mater. Chem. Front., 2017, 1, 1823–1828 RSC.
- Z. Li, Y. Wang, G. Baryshnikov, S. Shen, M. Zhang, Q. Zou, H. Ågren and L. Zhu, Nat. Commun., 2021, 12, 908 CrossRef CAS.
- M. Kohlhaas, M. Zähres, C. Mayer, M. Engeser, C. Merten and J. Niemeyer, Chem. Commun., 2019, 55, 3298–3301 RSC.
- D. Beaudoin, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2016, 55, 15599–15603 CrossRef CAS PubMed.
- N. L. C. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Y. Lam and B. Z. Tang, Chem. – Eur. J., 2014, 20, 15349–15353 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726–23740 RSC.
- H.-T. Feng, Y.-X. Yuan, J.-B. Xiong, Y.-S. Zheng and B. Z. Tang, Chem. Soc. Rev., 2018, 47, 7452–7476 RSC.
- Y. Ren, J. W. Y. Lam, Y. Dong, B. Z. Tang and K. S. Wong, J. Phys. Chem. B, 2005, 109, 1135–1140 CrossRef CAS PubMed.
- B.-P. Jiang, D.-S. Guo, Y.-C. Liu, K.-P. Wang and Y. Liu, ACS Nano, 2014, 8, 1609–1618 CrossRef CAS PubMed.
- N. Song, D.-X. Chen, Y.-C. Qiu, X.-Y. Yang, B. Xu, W. Tian and Y.-W. Yang, Chem. Commun., 2014, 50, 8231–8234 RSC.
- G. Markiewicz, A. Jenczak, M. Kołodziejski, J. J. Holstein, J. K. M. Sanders and A. R. Stefankiewicz, Nat. Commun., 2017, 8, 15109 CrossRef PubMed.
- F. Perlitius, A. Walczak, M. Čonková, G. Markiewicz, J. Harrowfield and A. R. Stefankiewicz, J. Mol. Liq., 2022, 367, 120511 CrossRef CAS.
- Y. Shishido, H. Anetai, T. Takeda, N. Hoshino, S.-I. Noro, T. Nakamura and T. Akutagawa, J. Phys. Chem. C, 2014, 118, 21204–21214 CrossRef CAS.
- H. Anetai, Y. Wada, T. Takeda, N. Hoshino, S. Yamamoto, M. Mitsuishi, T. Takenobu and T. Akutagawa, J. Phys. Chem. Lett., 2015, 6, 1813–1818 CrossRef CAS PubMed.
- W. Jiang, K. Tiefenbacher, D. Ajami and J. Rebek, Chem. Sci., 2012, 3, 3022–3025 RSC.
- B. Stuart, Infrared spectroscopy: fundamentals and applications, J. Wiley, Chichester, West Sussex, England, 2004 Search PubMed.
- T. Takeda, S. Yamamoto, M. Mitsuishi and T. Akutagawa, Org. Biomol. Chem., 2016, 14, 8922–8926 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on synthesis and Characterisation. See DOI: https://doi.org/10.1039/d3cc01263h |
| ‡ These authors contributed equally in this work. |
| This journal is © The Royal Society of Chemistry 2023 |