
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assembly of split aptamers by dynamic pH-responsive covalent ligation†
Aapo
Aho
 and
Pasi
Virta
*
and
Pasi
Virta
*
Department of Chemistry, University of Turku, Henrikinkatu 2, 20500 Turku, Finland. E-mail: pamavi@utu.fi
First published on 13th April 2023
Abstract
Reversible pH-responsive N-methoxyoxazolidine formation is used to ligate split aptamer fragments. Two twice-split models and one thrice-split model of CBA (cocaine-binding aptamer) were examined. The aptamer assembly was dynamic, proportional to the substrate concentration and occurred without interfering background ligation.
Aptamers, engineered by the SELEX technique1 for a variety of small and macromolecular targets, are high affinity binders which have received recognized value in therapeutic and diagnostic applications.2–8 Aptamers consisting of two or more fragments can still maintain sufficient recognition characteristics. These so-called split aptamers have caught attention as biosensors.9,10 The concept is based on the increased local concentration of split aptamer fragments, due to the concomitant binding to the target substrate. The proximity of the split sequences may be designed to initiate a signal, such as fluorescence or catalytic activity.11–17 In essence, such a split aptamer is a molecular switch, which may be integrated as a part of a cascade for higher functions. Splitting of an aptamer, while retaining the affinity and the specificity to the target, is challenging and only a small margin of known aptamers has been split successfully for this purpose.9,18 The affinity can be enhanced by co-operatively formed covalent linkages. This has been demonstrated in cocaine detection using strain-promoted alkyne-azide cycloaddition (SPAAC)12,19 or reductive amination20 between the split sequences of the cocaine binding aptamer (CBA). These rapid and irreversible ligation reactions raise the problem of non-specific aptamer assembly and hamper the reversibility of the target-induced signalling. Careful control of reaction times is required for meaningful monitoring of such systems. So far, there have been no reports on the dynamic covalent ligation of split aptamers, albeit reusable biosensors and dynamic systems21–29 alike could benefit from it.
Recently we showed that N-methoxyoxazolidines (1) form between 2′-deoxy-2′-N-(methoxyamino)uridine (2) and an aldehyde (Scheme 1) with an equilibrium constant of up to K = 9000 M−1 under aqueous conditions.30,31 Under acidic conditions the reaction is dynamic, but at pH 7 or above the products are stable and they can be isolated if needed. We also evaluated the applicability of the reaction for dynamic DNA-templated ligation.32 The DNA-template increased the rate and yield of the N-methoxyoxazolidine formation ca. 570- and 140-fold respectively compared to a non-templated one. Encouraged by these preliminary experiments, we herein demonstrate that this dynamic and pH-responsive reaction can be applied for reversible assembly of split aptamers. This is the first description of a dynamic ligation between split aptamer fragments utilizing a small-molecule substrate as a template.
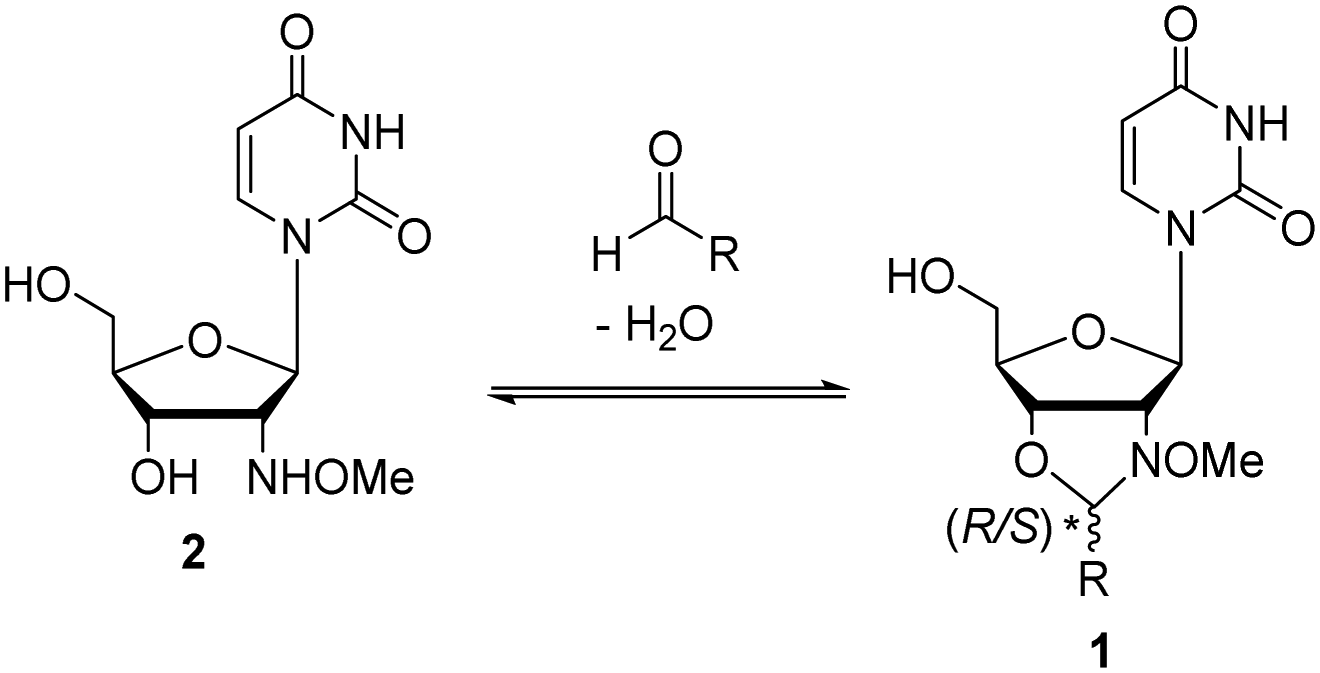 | ||
| Scheme 1 N-methoxyoxazolidine (1) formation between nucleoside 2 and an aldehyde. | ||
Aldehyde- and 2′-deoxy-2′-N-(methoxyamino)uridine-modified oligonucleotide (ON) fragments of the CBA and of model ONs were first synthesized. As we recently32 demonstrated with DNA-templated pseudo trans-N,O-acetalization systems, N-methoxyoxazolidine-protected ONs are convenient precursors for aldehyde-modified ONs. Therefore, dinucleosidic phosphoramidite 3 was prepared and used for the automated synthesis of ONs 1–4, outlined in Scheme 2. The key intermediate 4 (3′-TBDMS-5′-deoxy-5′-(2-oxoethyl)thymidine37) was condensed with 2 to yield N-methoxyoxazolidine UT dimer 5. The 5′-OH of 5 was acetylated, the 3′-O-TBDMS protection was removed and the exposed 3′-OH (6) was phosphitylated to obtain phosphoramidite 3, which was used for the automated DNA-synthesis. ON1–ON3 were synthesized on a regular DNA solid support and 2′-deoxy-2′-N-(methoxyamino)uridine-modified ONs (ON4–ON7) on a customized30 solid support (7). After the chain elongation, the ONs were released using concentrated aqueous ammonia (55 °C, 16 h) to obtain N-methoxyoxazolidine protected 5′-aldehyde-ONs (ON1–ON3), 2′-deoxy-2′-N-(methoxyamino)uridine-modified ONs (ON5–ON7) and one bifunctional ON (ON4).
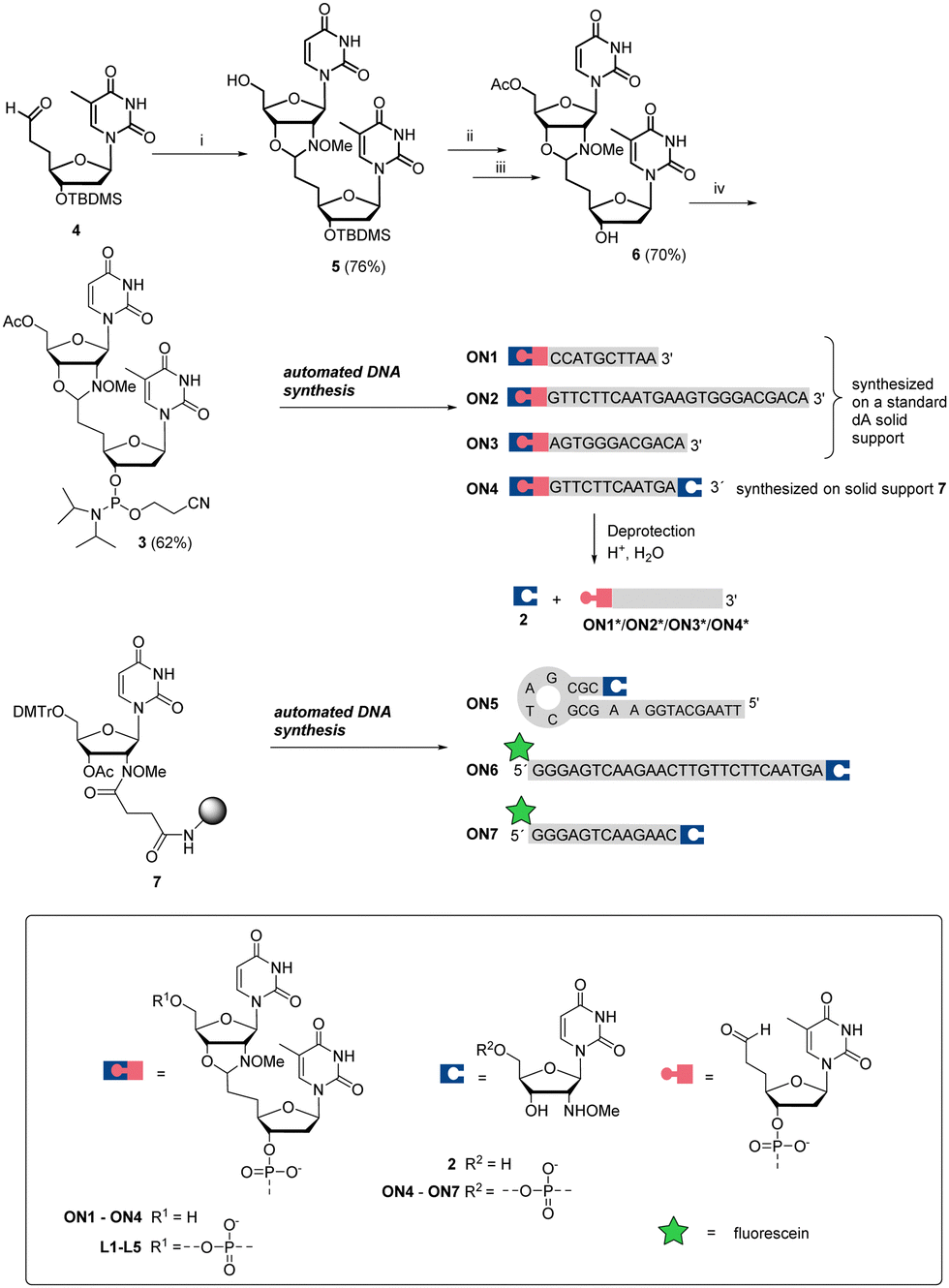 | ||
| Scheme 2 The synthesis of phosphoramidite 3,5′-aldehydic ONs (including protected aldehydes ON1, ON2, ON3, and ON4 and deprotected free aldehydes ON1*, ON2*, ON3*, and ON4*) and UNOMe-ONs (ON5, ON6, and ON7). Reaction conditions: (i) nucleoside 2 (1.0 eq.), acetic acid (25%, v/v) in DMSO; (ii) acetic anhydride, DMAP, pyridine; (iii) tetrabutylammonium fluoride, THF; (iv) N,N-diisopropylaminecyanoethylchlorophosphoramidite, acetonitrile. The symbolic expressions denoted here are used throughout the text. | ||
Prior to split aptamer studies, the dynamism and reaction kinetics of the N-methoxyoxazolidine ligation were examined by LC/MS analysis of simple model reactions using ON1, ON4 and ON5 (further details in the ESI†). The hydrolysis of the N-methoxyoxazolidine cap of ON1 followed reversible bimolecular reaction kinetics with pH-dependent half-life (t0.5) of 8.57 h to 70 d at pH 4 to 7 at r.t. Practically quantitative cleavage of the protection group (2) was observed, stalling at >96% of ON1* at equilibrium. When an excess of 2 (500 μM, 100 eq.) was added into the reaction mixture, the equilibrium shifted back to ca. 32% of ON1. In a similar experiment, bifunctional ON4* existed primarily as a cyclic N-methoxyoxazolidine-linked form and formed only a trace amount of ON4 when the same excess of 2 was added. When complementary ON5 (5 μM, 1 eq.) was added into the deprotected mixture of ON1* (ca. 5 μM), the self-templated hairpin product L1 was obtained (Scheme 3) in ca. 78% equilibrium yield. The DNA-templated ligation was facile with t0.5 of 3.42 min to 27.9 h at pH 5 to 10. Finally, the L1 formation was studied in a pseudo trans-N,O-acetalization system by mixing the protected ON1 (5 μM) and ON5 (5 μM) without a preliminary deprotection step. The yield of L1 did not change, but the rate (t0.5 of 7.69 h to 9.5 d at pH 4 to 6) was limited by the deprotection step. As a conclusion, the N-methoxyoxazolidine ligation and the masked aldehyde ONs proved suitable for dynamic hybridization-driven DNA-templated reactions potentially applicable for split aptamers.
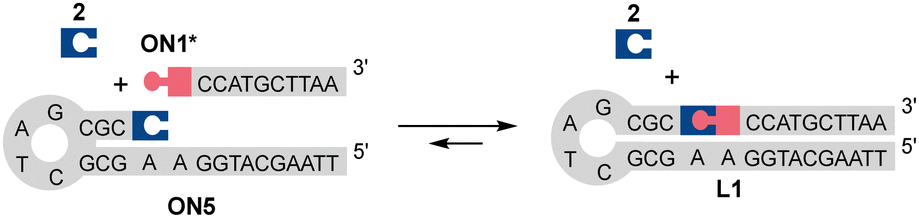 | ||
| Scheme 3 DNA-templated ligation between ON1* (ca. 5 μM) and ON5 (5 μM) in the presence of the cleaved protecting group 2 (ca. 5 μM). The symbolic expressions are denoted in Scheme 2. | ||
The dynamic covalent aptamer assembly was then studied using split CBA models. CBA has been characterized to bind to the substrate as a three-way junction with two loops.38,39 The ligation sites were placed in the loop regions of CBA, which are likely to be more tolerant of structural modifications and allow more freedom for the formation of variable R/S-isomers of the N-methoxyoxazolidine linkage (in comparison to the potential sites in the stem regions, if applied). First, we examined two split CBA models: A and B, in which the CBA was split into two fragments. These fragments were elongated by 3′-UNOMe, 5′-fluorescein, and 5′-aldehyde accordingly (Scheme 4). A is similar to a model that has previously been studied in noncovalent16 and covalent12 aptamer assemblies, while model B has an alternative splitting site. In both models, quinine was used as the ligation-inducing substrate (the CBA binds to quinine with comparable affinity to cocaine).38,40,41 Split sequences (ON7 with ON2 and ON6 with ON3, 1 μM each), representing these models, were incubated with variable amount of quinine (0, 10, 100, or 1000 μM) in aqueous solution adjusted to pH 5 and the mixtures were monitored by denaturing PAGE and mass spectrometry. To ensure complete deprotection of the aldehyde, the mixtures were first incubated for 24 h at 55 °C. At this point, neither of the models showed any ligation product. To initiate the aptamer assembly, the mixtures were cooled down and incubated at room temperature. After one hour, model A showed 1.6–72% yield of L2 in the presence of 0–1000 μM of quinine. The assembly stalled to 18–89% equilibrium of L2 after 24 h, which persisted after one month of incubation (Fig. 1). We also examined a control reaction of model A, in which ON7 or ON2 was replaced with a regular DNA (see the ESI†). As expected, no product formation was observed. In contrast to model A, model B showed only 0–22% of L3 after one day and stalled to equilibrium of 1.5–36% after one week in the presence of 10–1000 μM of quinine. In contrast to model A, model B did not yield any substrate-independent ligation product (0 μM quinine) even after one month (Fig. 1). While formation of L2 is more favored than that of L3, the covalent aptamer assembly is driven by complexation to quinine in both models both kinetically and thermodynamically. After one month of incubation at room temperature, the reaction mixtures of model A were again heated to 55 °C for 24 h and then cooled back to RT. The ligation product L2 was disassembled completely by the heating step and then reassembled with similar yields to the first assembly (see details in the ESI†). Thus, the assembly was truly reversible.
 | ||
| Scheme 4 Split aptamer models A, B, and C. The symbolic expressions are denoted in Scheme 2. | ||
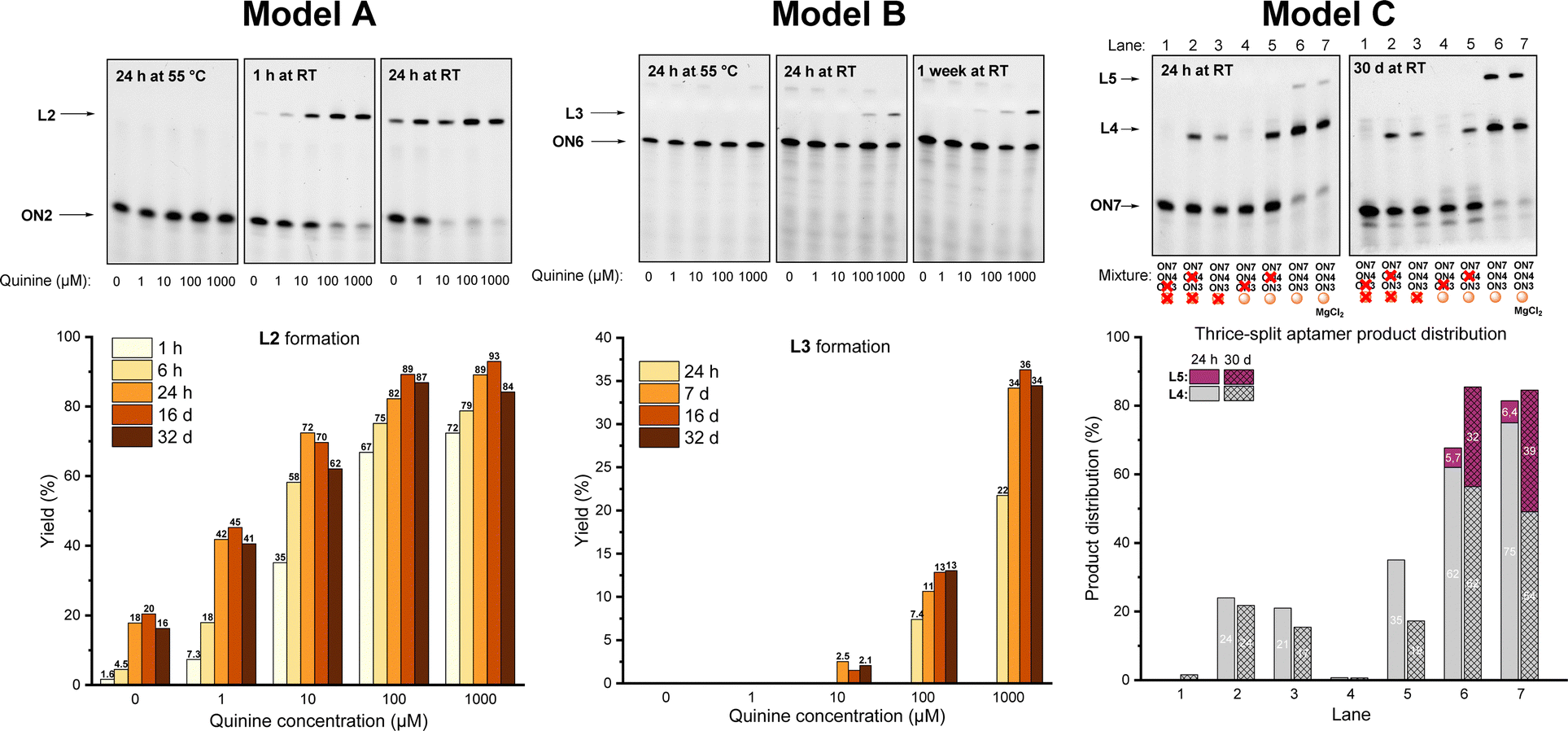 | ||
| Fig. 1 Denaturing PAGE analysis and ligation yield in split aptamer models A, B, and C. See the ESI† for mass spectra of L2, L3, L4 and L5. | ||
The limitations of the aptamer assembly were further examined by implementing both split sites in thrice split model C using UNOMe- and aldehydic-ONs (ON7 and ON3) and a bifunctional one (ON4) to form full aptamer L5 (Scheme 4). The split fragments ON7, ON3, and ON4 (5 μM of each) were incubated with quinine (100 μM) in aqueous solution (pH 5) overnight at 55 °C, cooled down and then incubated at room temperature. After one day, 72% of truncated product L4 and 5.7% of full aptamer L5 were formed (Fig. 1, lane 6). The yield of aptamer L5 rose to 32% after one month. In parallel with the main reactions, multiple exclusion experiments were run, in which one of the ONs or quinine was removed. No L5 was found even after one month if quinine was excluded from the mixture (Fig. 1, lane 3). Without quinine, the yield of L4 also dropped (from 72% to 21% after 24 h, cf. lanes 6 and 3 in Fig. 1). This does not necessarily mean that L4 is a functional aptamer itself, as removal of ON4 also reduced the yield of L4 (Fig. 1, lane 5). The role of Mg2+ (50 mM MgCl2) for the assembly was examined. A slight improvement in the yield of L5 (39% after one month) was observed.
In summary, split aptamer fragments can be reversibly ligated via pH-controlled N-methoxyoxazolidine linkage. The N-methoxyoxazolidine ligation between the CBA fragments was found to be dynamic and highly dependent on the quinine concentration. Of the twice split models, model A performed better in terms of the reaction rate and yield. The fidelity of model A was robust with ca. 2-fold higher aptamer formation in the presence of 1 μM of quinine compared to background (0 μM quinine). More importantly, this selectivity was observed in the equilibrated reaction mixture. In addition to the thermodynamic stabilization, quinine enhanced the rate of aptamer ligation. Therefore, higher fidelity can be obtained by optimization of the monitoring time. The aptamer ligation could be reversed by heat–cool cycles, underlining the dynamicity. Twice split model B was slower and required more quinine to form the product, but the fidelity was exceptional with zero background at the putative equilibrium state. Similarly, the thrice split model C produced zero background. Noteworthily, these experiments were performed at pH 5, whereas DNA hybridizes most efficiently around pH 7. Due to partial complementary nature of the CBA fragments, higher ligation yields may be obtained by increasing the pH of the reaction solution. Depending on the substrate–aptamer interactions, the fidelity may also be altered by pH change. Experiments regarding the formation of hairpin L1 showed that N-methoxyoxazolidinization proceeds well in a remarkable wide pH range (4–10) if a suitable template is used, which leaves room for pH optimization. Based on these findings, the N-methoxyoxazolidine ligation proved its applicability for dynamic covalent assembly of split aptamers, which can be isolated on demand. In a wider perspective, N-methoxyoxazolidine formation was shown to be a considerable tool for target-induced dynamic combinatorial chemistry, for which a new type of biocompatible and conditionally reversible reaction has been called for.28,33–36
Aapo Aho: conceptualization, methodology, investigation, formal analysis, and writing. Pasi Virta: conceptualization, methodology, and writing.
A. A acknowledges Doctoral Programme in Exact Sciences (Exactus), University of Turku. P. V. acknowledges the Academy of Finland project (308931).
Conflicts of interest
There are no conflicts to declare.References
- M. Kohlberger and G. Gadermaier, Biotechnol. Appl. Biochem., 2022, 69, 1771 CrossRef CAS PubMed.
- J. Zhou and J. Rossi, Nat. Rev. Drug Discovery, 2016, 16, 181 CrossRef PubMed.
- S. M. Nimjee, R. R. White, R. C. Becker and B. A. Sullenger, Annu. Rev. Pharmacol. Toxicol., 2017, 57, 61 CrossRef CAS PubMed.
- P. K. Kulabhusan, B. Hussain and M. Yüce, Pharmaceutics, 2020, 12, 1 Search PubMed.
- T. H. Ku, T. Zhang, H. Luo, T. M. Yen, P. W. Chen, Y. Han and Y. H. Lo, Sensors, 2015, 15, 16281 CrossRef CAS PubMed.
- W. Zhou, Jimmy Huang, P. J. Ding and J. Liu, J. Analyst., 2014, 139, 2627 RSC.
- H. Yu, O. Alkhamis, J. Canoura, Y. Liu and Y. Xiao, Angew. Chem., Int. Ed., 2021, 60, 16800 CrossRef CAS PubMed.
- S. Ng, H. S. Lim, Q. Ma and Z. Gao, Theranostics, 2016, 6, 1683 CrossRef CAS PubMed.
- M. Debiais, A. Lelievre, M. Smietana and S. Müller, Nucleic Acids Res., 2020, 48, 3400 CrossRef CAS PubMed.
- A. Chen, M. Yan and S. Yang, TrAC, Trends Anal. Chem., 2016, 80, 581 CrossRef CAS.
- S. Zhang, K. Wang, J. Li, Z. Li and T. Sun, RSC Adv., 2015, 5, 75746 RSC.
- A. K. Sharma, A. D. Kent and J. M. Heemstra, Anal. Chem., 2012, 84, 6104 CrossRef CAS PubMed.
- J. L. He, Z. S. Wu, H. Zhou, H. Q. Wang, J. H. Jiang, G. L. Shen and R. Q. Yu, Anal. Chem., 2010, 82, 1358 CrossRef CAS PubMed.
- J. Zhu, L. Zhang, Z. Zhou, S. Dong and E. Wang, Anal. Chem., 2014, 86, 312 CrossRef CAS PubMed.
- S. Chi-Chin Shiu, Y.-W. Cheung, R. M. Dirkzwager, S. Liang, A. B. Kinghorn, L. A. Fraser, M. S. L. Tang, J. A. Tanner, S. Chi-Chin Shiu, Y. Cheung, R. M. Dirkzwager, S. L. Liang, A. B. Kinghorn, L. A. Fraser, M. S. L. Tang and J. A. Tanner, Adv. Biosyst., 2017, 1, 1600006 CrossRef PubMed.
- M. N. Stojanovic, P. de Prada and D. W. Landry, J. Am. Chem. Soc., 2000, 122, 11547 CrossRef CAS PubMed.
- C. H. Lu, F. Wang and I. Willner, Chem. Sci., 2012, 3, 2616 RSC.
- R. Wang, Q. Zhang, Y. Zhang, H. Shi, K. T. Nguyen and X. Zhou, Anal. Chem., 2019, 91, 15811 CrossRef CAS PubMed.
- A. K. Sharma and J. M. Heemstra, J. Am. Chem. Soc., 2011, 133, 12426 CrossRef CAS PubMed.
- N. G. Spiropulos and J. M. Heemstra, Artif. DNA PNA XNA, 2012, 3, 123 CrossRef PubMed.
- P. Frei, R. Hevey and B. Ernst, Chem. – Eur. J., 2019, 25, 60 CrossRef CAS PubMed.
- F. B. L. Cougnon and J. K. M. Sanders, Acc. Chem. Res., 2012, 45, 2211 CrossRef CAS PubMed.
- F. v Reddavide, W. Lin, S. Lehnert and Y. Zhang, Angew. Chem., Int. Ed., 2015, 54, 7924 CrossRef CAS PubMed.
- E. Moulin, G. Cormos and N. Giuseppone, Chem. Soc. Rev., 2012, 41, 1031 RSC.
- B. C. Buddingh’ and J. C. M. van Hest, Acc. Chem. Res., 2017, 50, 769 CrossRef PubMed.
- G. Clixby and L. Twyman, Org. Biomol. Chem., 2016, 14, 4170 RSC.
- P. Adamski, M. Eleveld, A. Sood, Á. Kun, A. Szilágyi, T. Czárán, E. Szathmáry and S. Otto, Nat. Rev. Chem., 2020, 4, 386 CrossRef.
- J. Li, P. Nowak and S. Otto, J. Am. Chem. Soc., 2013, 135, 9222 CrossRef CAS PubMed.
- A. Herrmann, Chem. Soc. Rev., 2014, 43, 1899 RSC.
- A. Aho, A. Äärelä, H. Korhonen and P. Virta, Molecules, 2021, 26, 490 CrossRef CAS PubMed.
- A. Aho, M. Sulkanen, H. Korhonen and P. Virta, Org. Lett., 2020, 22, 6714 CrossRef CAS PubMed.
- A. Aho, T. Österlund, J. Rahkila and P. Virta, Eur. J. Org. Chem., 2022, 8–13 Search PubMed.
- L. Monjas and A. K. H. Hirsch, Future Med. Chem., 2015, 7, 2095 CrossRef CAS PubMed.
- M. Mondal and A. K. H. Hirsch, Chem. Soc. Rev., 2015, 44, 245 RSC.
- N. Maraković and G. Šinko, Acta Chim. Slov., 2017, 64, 15 Search PubMed.
- S. Otto, R. L. E. Furlan and J. K. M. Sanders, Curr. Opin. Chem. Biol., 2002, 6, 321 CrossRef CAS PubMed.
- T. J. Caulfield, C. V. C. Prasad, C. P. Prouty, A. K. Saha, M. P. Sardaro, W. C. Schairer, A. Yawman, D. A. Upson and L. I. Kruse, Bioorg. Med. Chem. Lett., 1993, 3, 2771 CrossRef CAS.
- A. A. Shoara, S. Slavkovic, L. W. Donaldson and P. E. Johnson, Can. J. Chem., 2017, 95, 1253 CrossRef CAS.
- M. A. D. Neves, O. Reinstein and P. E. Johnson, Biochemistry, 2010, 49, 8478 CrossRef CAS PubMed.
- R. Pei, A. Shen, M. J. Olah, D. Stefanovic, T. Worgall and M. N. Stojanovic, Chem. Commun., 2009, 3193 RSC.
- M. N. Stojanovic and T. S. Worgall, Curr. Opin. Chem. Biol., 2010, 14, 751 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on synthesis and kinetic experiments, NMR and mass spectrometric data. See DOI: https://doi.org/10.1039/d3cc01158e |
| This journal is © The Royal Society of Chemistry 2023 |