
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA photochromic metallacycle with highly anisotropic Dy–F magnetic units†
Nour
El Beyrouti
a,
Felix
Houard
 a,
Marie
Cordier
a,
Elzbieta
Trzop
b,
Stéphane
Rigaut
a,
Boris
Le Guennic
a,
Kevin
Bernot
a and
Lucie
Norel
*a
a,
Marie
Cordier
a,
Elzbieta
Trzop
b,
Stéphane
Rigaut
a,
Boris
Le Guennic
a,
Kevin
Bernot
a and
Lucie
Norel
*a
aUniv Rennes, INSA Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, F-35000 Rennes, France. E-mail: lucie.norel@univ-rennes1.fr
bUniv Rennes, CNRS, IPR (Institut de Physique de Rennes) – UMR 6251, F-35000 Rennes, France
First published on 22nd March 2023
Abstract
A dinuclear metallacycle assembled from a bispyridyl dithienylethene linker and a highly anisotropic dysprosium based Single Molecule Magnet (SMM) shows magnetic hysteresis at 1.8 K together with photoisomerization in single crystals (SC). The impact of photoswitching on the SMM behavior is evidenced and related to the specific organization of the magnetic units.
The large magnetic anisotropy exhibited by specific lanthanide complexes is the origin of SMM behaviour, characterized by long relaxation times of the magnetization and eventually magnetic hysteresis at temperatures ranging from liquid helium to liquid nitrogen.1 Such a property, combined with the quantic nature of the complexes opens new possibilities in various fields such as spintronic.2 In addition, the molecular nature of these architectures makes the introduction of other functions straightforward, therefore, extending the scope of possible applications.3 For instance, the combination of SMM behaviour and light remote control can be targeted through simple association of an anisotropic magnetic building unit and a photoactive ligand,3b,c such as a photochromic dithienylethene derivative4 that can undergo reversible transformation between two, so called, open and closed isomers.
Recently, we have used such an approach to obtain magnetic hysteresis photomodulation within chains of lanthanide single-molecule magnets having Dy(Tppy)F (Tppy = tris(3-(2-pyridyl)pyrazolyl)hydroborate) magnetic units and bispyridyl terminated dithienylethene (DTE) photoswitches (compound Dy-1D in Fig. 1).5
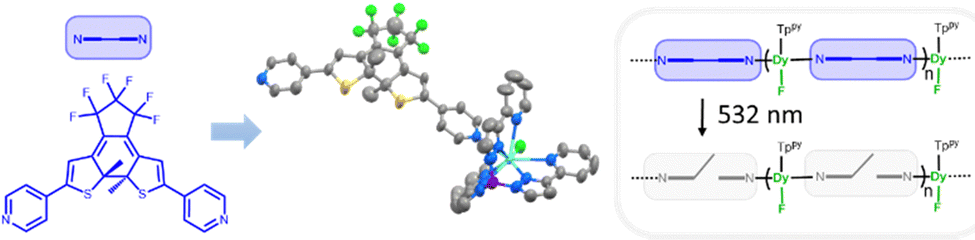 | ||
| Fig. 1 previously investigated Dy-1D compound. | ||
In the present work, we investigate how the synthetic versatility of this unprecedented approach can be exploited to generate other topologies and draw structure–property relationships concerning this photomagnetic effect. More specifically, in comparison with Dy-1D,5 we have used 1,2-bis(2-methyl,5-(3-pydidyl)-3-thienyl)perfluorocyclopentene,6L (Fig. 2), instead of the analogous 4-pyridyl terminated ligand. The result is a cyclic dinuclear coordination compound, able to isomerize in SC and with a photomagnetic response markedly different from Dy-1D despite a similar metal coordination sphere. This study thus underlines the importance of crystal packing and its modification under light irradiation as the main parameter triggering the changes in the magnetic hysteresis response.
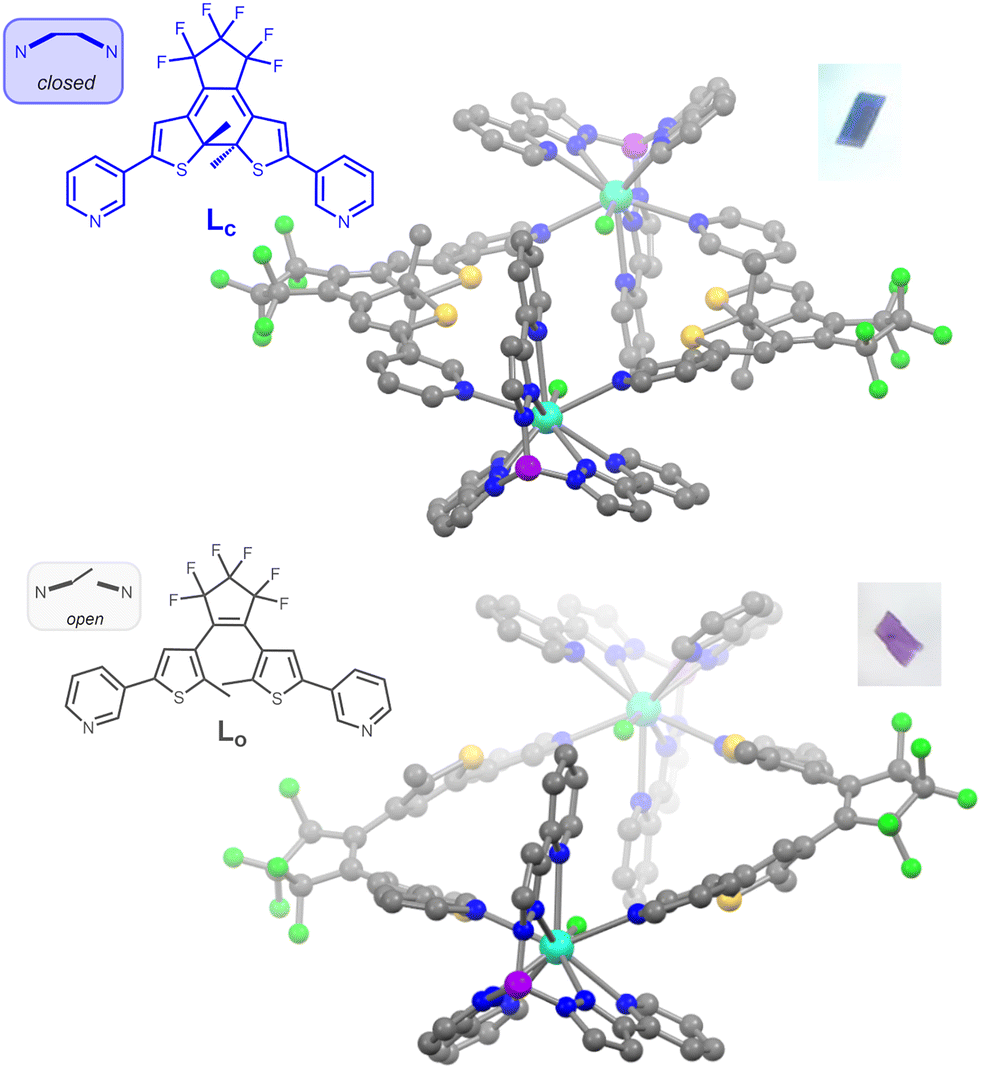 | ||
| Fig. 2 Views of the dicationic dinuclear metallacycles in compounds 1c (top, 150 K) and 1o (bottom, 90 K) as determined from single-crystal X-ray diffraction data. Grey, blue, red, purple, green, yellow and light blue spheres represent C, N, O, B, F, S and Dy atoms respectively; H atoms are omitted for clarity. Insets show photographs of the crystals. | ||
The title compound and its yttrium analogue were obtained by combination of the DTE ligand in its closed state Lc and [M(Tppy)F(pyridine)2]BArF (M = Y, Dy). These new precursors are easily synthesized by salt metathesis from the corresponding [M(Tppy)F(pyridine)2]PF6 complexes7 (see ESI†). They undergo the same exchange reaction of the coordinated pyridines with ditopic ligands, as previously obtained for Dy-1D,5 but the topology of the DTE linker induces here the formation of dinuclear rings rather than chains. The metallacycles obtained for both M = Dy (1c) and Y (2c), from slow evaporation of dichloromethane/hexane solutions, are isostructural (Table S3, ESI†). As a representative example, the X-ray structure of 1c reveals an asymmetric unit composed of one dysprosium atom coordinated to the Tppy ligand, one terminal fluoride atom and one pyridine group from the DTE ligand as well as a BArF anion. The ring structure is generated through an inversion center (space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) located in the center of the ring and disposes the two Dy–F bonds exactly antiparallel, with a Dy–Dy distance of 12.279 Å (Fig. 1). The cavity resulting from the ring formation is filled with residual electronic density (likely from solvent molecules) that we could not satisfyingly model.
) located in the center of the ring and disposes the two Dy–F bonds exactly antiparallel, with a Dy–Dy distance of 12.279 Å (Fig. 1). The cavity resulting from the ring formation is filled with residual electronic density (likely from solvent molecules) that we could not satisfyingly model.
As a result of ring formation, the geometry around the dysprosium ion is a 9 coordinated spherical capped square antiprism as shown by the SHAPE analysis,8 with a parameter of 0.863 for this geometry, consistent with the previous description of [Dy(Tppy)F(pyridine)2]PF6 (0.912)7 and Dy-1D (0.731).5 The main distortion to this ideal geometry comes from the very short Dy–F distance of 2.105(3) Å, which is expected to generate a strong axial magnetic anisotropy on each Dy center. The identification of bulk samples of 1c/2c as having the formula [(M(Tppy)F)2(Lc)2](BArF)2 is supported by elemental analysis, mass spectrometry, powder XRD data, as well as IR spectroscopy and, in the case of 2c, NMR spectroscopy.
Since dithienylethene undergo solid-state photochromism, we have studied the isomerization reaction in KBr pellets. A pellet containing 1c showed a deep blue color caused by a strong absorption band centered at 582 nm (Fig. 3). Together with the contribution at 374 nm, this band is the signature of the DTE linker in its closed state,6 whereas the DyTppyF unit is expected to absorb light only in the UV region of the spectrum. Upon white light irradiation for 90 min, the pellet became pale violet and the bands of the closed DTE are replaced by a weaker contribution with λmax = 552 nm in the photostationnary state (PSS). The initial blue color was restored upon irradiation at λ = 365 nm with a recovery of the initial spectrum of about 97% in intensity at 582 nm. Up to seven cycles of white light and UV exposure were successively performed without apparent fatigability due to degradation (Fig. S11, ESI†). The exact same behaviour was also observed for 2c (Fig. S10 and S12, ESI†). However, the full opening of a DTE linker5 under visible light irradiation is usually expected to lead to a colourless compound, and not the purple color obtained in the PSS.
To gain insights into this intriguing feature, TD-DFT calculations were performed on all possible isomers of the yttrium dinuclear structure (see ESI†). As expected, and after full geometry optimization (Fig. S30, ESI†), the calculated absorption spectrum for [(Y(Tppy)F)2(Lc)2]2+ is consistent with the experimental behaviour, and a transition with π–π* character is calculated at λmax = 609 nm (Table S14 and Fig. S31, ESI†). Similarly, the calculated spectrum for [(Y(Tppy)F)2(Lo)2]2+ has no absorption in the visible range. We also constructed and optimized the geometry of a dinuclear complex having only one closed isomer, with formula [(Y(Tppy)F)2(Lo)(Lc)]2+ and observed a shift of the main π–π* transition to higher energy with λmax = 602 nm (Table S14 and Fig. S32, ESI†), probably due to the distorsion of the conjugated path of the closed DTE upon the opening of the first ligand. Althoulgh this calculated shift is smaller than observed, we can tentatively assign the purple color to residual highly colored closed DTE units from a partially isomerized structure, influenced also by crystal packing inducing further distortion of the closed DTE.
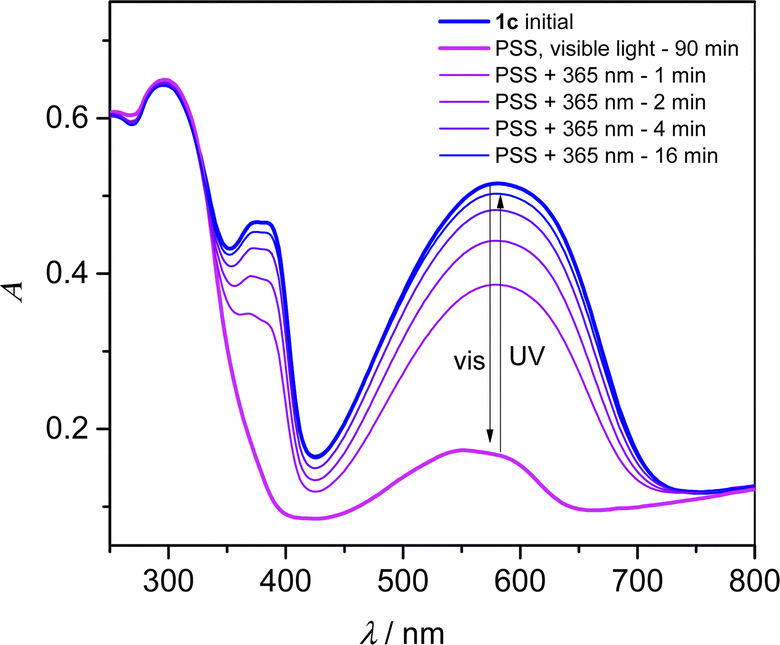 | ||
| Fig. 3 Electronic absorption spectra (KBr pellet) of 1c (blue line), the PSS after 90 min white light irradiation causing the opening reaction (purple line) and successively 1–16 min 365 nm irradiation causing the closing reaction. | ||
To further characterize the photoinduced open state, we first performed the isomerization reaction within SC. Upon 6h30 laser irradiation at λ = 660 nm (2.5 mW) of a batch of crystals of 1c, purple crystals were obtained and remained of sufficient integrity to allow for structural determination. The space group remains P after irradiation and the unit cell parameters show small variations upon light irradiation (Table S4, ESI†). The asymmetric unit is similar to the one of 1c but the DTE ligand is now in its open state (Fig. 1), as clearly seen from the opening of the 6-membered ring and the lengthening of the reactive carbon atoms distance from 1.51 to 3.38 Å. Thus, complete isomerization to the open isomer is observed in SC and the compound is named 1o (for “open”). Complementary evidence of full conversion comes from the 1H and 19F NMR analysis of a batch of 2c crystals, crushed and exposed to white light for 92 h until its absorption spectrum matches the one of the PSS. At this stage, the compound was dissolved in CD3OD, causing decoordination and formation of [Y(Tppy)F(CD3OD)2]+, BArF− and free DTE ligand (Fig. S16–S18, ESI†). The spectral signature of the later clearly indicates the presence of the open form with less than 1% of the closed form remaining after irradiation. Thus the PSS corresponds to an almost fully isomerized ring structure with formula [(M(Tppy)F)2(Lo)2](BArF)2 for 1o (M = Dy) and 2o (M = Y). Finally, only the opening reaction could be followed on SC but not the subsequent closing reaction since UV irradiation reduced the sample crystallinity.
Since our working hypothesis for SMM photo-modulation is that the crystal packing modification upon isomerisation can influence the magnetic properties, it is important to compare in details the structures of 1c and 1o. Concerning the dysprosium coordination sphere, it stays almost perfectly identical upon isomerization, as shown by the SHAPE parameter of 0.754 for 1o for the spherical capped square antiprism geometry and the overlay of the two coordination spheres (Fig. S4, ESI†). The Dy–F distance in 1o is the same as in 1c within experimental error (2.107(4) Å). We thus expect that crystal field splitting will be the same for 1c and 1o and will be comparable to the one observed for [Dy(Tppy)F(pyridine)2]PF67 or Dy-1D,5 with a total splitting of 800 cm−1 and a first excited state at around 300 cm−1. The Dy-Dy distance within the ring is larger in 1o with 13.079 Å (+0.80 Å) and the antiparallel disposition of the Dy–F dipoles persists (Fig. S5, ESI†). Actually, the closest Dy–Dy distance is the result of π stacking existing between adjacent pyrazolylpyridine arms and varies only slightly from 8.624 Å in 1c to 8.630 Å in 1o (Fig. S6, ESI†). The next closest Dy–Dy distance of 10.710 Å in 1c becomes shorter in 1o with 10.376 Å (−0.334 Å). These distances correspond to perfect antiparallel disposition of the Dy–F dipoles and thus potential antiferromagnetic dipolar couplings contrary to Dy-1D where ferromagnetic interactions were calculated.
The rings in 1o are heterochiral with two DTE with opposite helicity (M and P), since the homochiral arrangement is not compatible with the cyclic arrangement. Since each helicity of the open DTE is stereoselectively obtained from a given disposition of the methyl groups in the closed state, with for instance the M helix resulting from the opening of the (S,S) configuration of the chiral carbon centers, it means that starting closed compound 1c also shows heterochiral (R,R), (S,S) ring structures, probably induced by the crystalisation process from a mixture of several stereoisomers in dynamic equilibrium. Indeed, the inversion center in 1c imposes the heterochiral disposition of the DTE even if the methyl groups of the DTE show substantial crystallographic disorder modelled with 3 possible dispositions roughly corresponding to the two enantiomers of the DTE in a 47.9 to 52.1% ratio.
Compounds 1c and 1o were then characterized in the solid state by static (dc) and dynamic (ac) magnetization measurements (Fig. S22–S29 and Table S5–S13, ESI†). The room temperature χMT values for both 1c and 1o (28.30 and 28.50 emu K mol−1, respectively) are close to the predicted value of 28.34 emu K mol−1 for a dinuclear system with isolated Dy3+ ions (6H15/2, S = 5/2, L = 5, and g = 4/3) and are in excellent agreement with previously investigated compounds of this family. These χMT values start to decrease steadily below 200 K, before dropping dramatically below 3.5 K mainly because of saturation effect.
| τ−1 = BTn + τ0−1 exp(−Ueff/kBT) + τtunnel−1 | (1) |
The ac magnetic susceptibility measurements for both compounds exhibited peaks in the out-of-phase susceptibility (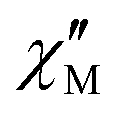 ) vs. frequency curves at temperatures up to 46 K, in the absence of an applied dc field. The low temperature peaks exhibit little temperature dependence and as a result the Arrhenius plot (Fig. 4) of the relaxation times versus inverse temperature shows a plateau at the lowest temperature. This behaviour is consistent with quantum tunnelling as the primary magnetic relaxation pathway in the 2–6 K range for both compounds but with a slightly faster relaxation in the case of 1o compared with 1c (0.495 s and 0.140 s at 2 K respectively). At higher temperature, a shift of the out-of-phase peaks with temperature is observed, consistent with Raman and Orbach relaxation processes taking over upon rising the temperature. It is striking that the relaxation time for 1o and 1c tend to merge when the temperature is above 20 K meaning that the Orbach regime is common to both compounds. Actually, upon application of a dc field of 1600 Oe, which was determined to be optimal for slowing down the magnetic relaxation for both compounds, the data collected in the 0.7–1500 Hz window are nearly identical (within error) for 1c and 1o, underlying that the main difference in their behaviour is the quantum tunnelling rate. Therefore, we fitted the temperature dependence of the relaxation time using the eqn (1) which includes Raman, Orbach and quantum tunnelling contributions.
) vs. frequency curves at temperatures up to 46 K, in the absence of an applied dc field. The low temperature peaks exhibit little temperature dependence and as a result the Arrhenius plot (Fig. 4) of the relaxation times versus inverse temperature shows a plateau at the lowest temperature. This behaviour is consistent with quantum tunnelling as the primary magnetic relaxation pathway in the 2–6 K range for both compounds but with a slightly faster relaxation in the case of 1o compared with 1c (0.495 s and 0.140 s at 2 K respectively). At higher temperature, a shift of the out-of-phase peaks with temperature is observed, consistent with Raman and Orbach relaxation processes taking over upon rising the temperature. It is striking that the relaxation time for 1o and 1c tend to merge when the temperature is above 20 K meaning that the Orbach regime is common to both compounds. Actually, upon application of a dc field of 1600 Oe, which was determined to be optimal for slowing down the magnetic relaxation for both compounds, the data collected in the 0.7–1500 Hz window are nearly identical (within error) for 1c and 1o, underlying that the main difference in their behaviour is the quantum tunnelling rate. Therefore, we fitted the temperature dependence of the relaxation time using the eqn (1) which includes Raman, Orbach and quantum tunnelling contributions.
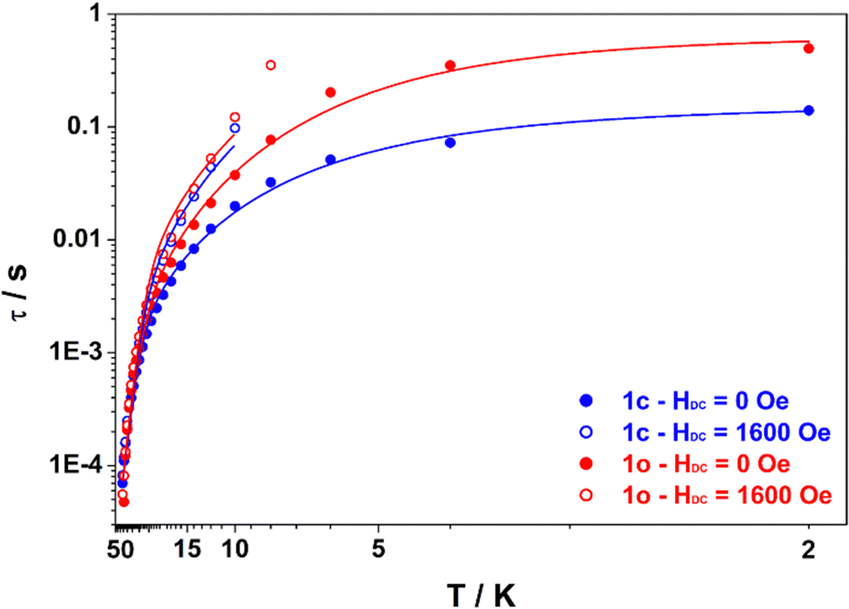 | ||
| Fig. 4 Plot of the relaxation time τ (log scale) versus T (inverse scale) for 1c (blue) and 1o (red) measured under HDC = 0 Oe (full dots) and HDC = 1600 Oe (open dots). Lines are best fits according to eqn (1) assuming fixed values of Ueff = 193 cm−1 and τ0 = 1.86 × 10−7 s for both compounds with the parameters from Table 1 (0 Oe) and Table S13 (ESI†) (1600 Oe). | ||
In line with our observation, unique parameters where used to fit the Orbach regime for both compounds in both applied fields. The quantum tunnelling parameters were fixed to zero for 1600 Oe applied field and Raman parameters were optimized in all cases. The result of this fitting procedure is shown in Table 1 (0 Oe) and Table S13 (ESI†) (1600 Oe). The relaxation barrier of 193 cm−1 agrees well with the ones previously determined for compounds with similar coordination spheres (like Dy-1D) and are probably underestimated by the fitting procedure since ab initio calculations predict 282 cm−1 for a virtually identical coordination sphere in [Dy(Tppy)F(pyridine)2]PF6.7 This barrier remains constant upon isomerization in agreement with the coordination sphere upholding (see supra). Finally, the major effect of isomerization is the slowing down of quantum tunneling roughly by a factor of 4 and overall, these parameters make the relaxation behaviour of both 1c and 1o slower than that of Dy-1D for which the limiting tunnelling time was at best 0.0337 s. Variable-field magnetization data collected for 1c and 1o at 1.8 K revealed the presence of butterfly magnetic hysteresis similar to that observed for previous complexes of this family (Fig. 5).5 As expected from the limited changes in the different parameters describing the relaxation processes, the hysteresis curves of 1c and 1o show pretty much the same characteristics and unlike the behavior of Dy-1D, the DTE opening does not cause a shrinking of the hysteresis loop.
| 1c | 1o | |
|---|---|---|
| U eff (cm−1) | 193 (±17) | |
| τ 0 (10−7 s) | 1.86 (±0.35) | |
| τ QT (s) | 0.16 (±0.03) | 0.66 (±0.21) |
| B (s−1 K−n) | 0.22 (±0.08) | 0.030 (±0.016) |
| n | 2.36 (±0.12) | 2.90 (±0.19) |
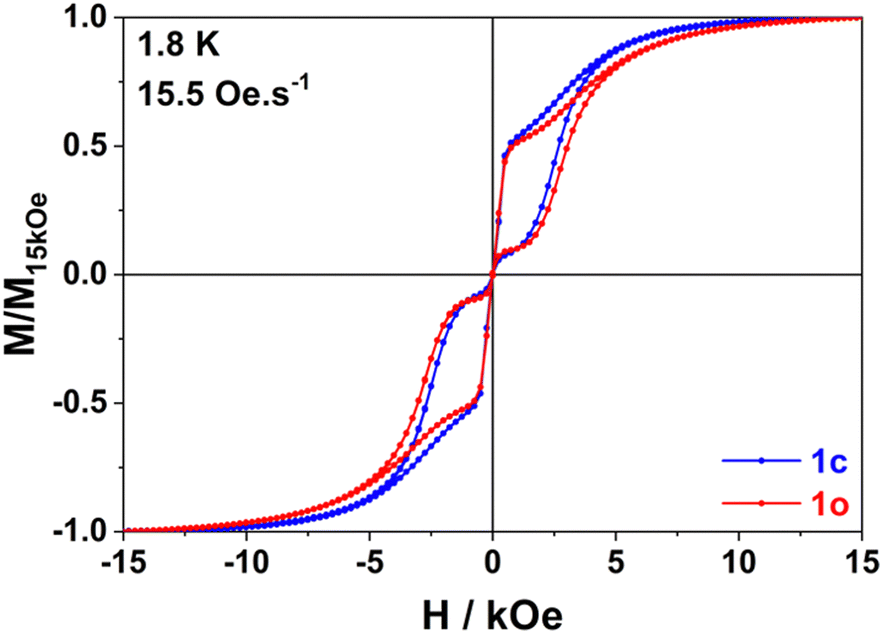 | ||
| Fig. 5 Variable field magnetization data for 1c (blue) and 1o (red) collected at 1.8 K using a field sweep rate of 15.5 Oe min−1. Data are normalized on the basis of the magnetization value measured at 15 kOe. | ||
We link hysteresis persistence under light-switching to several parameters. First, the unchanged coordination sphere of the dysprosium ion lead to the same energy barrier for the Orbach process. Second, the Raman parameters are not dramatically changed. However, these two observations also hold for Dy-1D and this compound clearly shows larger changes in its hysteresis. So, the main difference between these two cases comes from the reduced influence of the opening reaction on quantum tunnelling (÷22 in Dy-1D, ×4 in 1) that can be tentatively linked with the changes in dipolar couplings. Indeed, for 1 the shorter Dy–Dy distance is almost unchanged during the isomerization process (+0.006 Å) contrary to what is seen on Dy-1D (−0.107 Å). This means that the main contribution to the change in dipolar coupling is here cancelled.
To conclude, our versatile synthetic approach allows the synthesis of chains as well as unprecedented ring structures having two anisotropic dysprosium centers linked by DTE units. The linker opening reaction proceed in SC with full conversion and crystal structure was determined on both photo-isomers. However, unlike its chain analogue, this is not accompanied by important changes in the slow relaxation dynamics, as a result of the specific packing and arrangement of dysprosium atoms in this structure that ensure a very robust magnetic behaviour, with only marginal modification of dipolar interactions. We are currently investigating how other photochromic switches can lead to coordination compounds combining crystal tolerance to photoisomerization with significant changes in their magnetic behaviour.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) C. Benelli and D. Gatteschi, Introduction to Molecular Magnetism, From transition metals to lanthanides, Wiley-VCH, Weinheim, Germany, 2015 Search PubMed; (b) K. L. M. Harriman, D. Errulat and M. Murugesu, Trends Chem., 2019, 1, 425–439 CrossRef CAS; (c) N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed; (d) F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362(6421), 1400 CrossRef CAS PubMed; (e) C. A. P. Goodwin, Dalton Trans., 2020, 49, 14320–14337 RSC.
- (a) S. Sanvito, Molecular spintronics, Chem. Soc. Rev., 2011, 40, 3336–3355 RSC; (b) E. Coronado, Nat. Rev. Mater., 2020, 5, 87–104 CrossRef.
- (a) O. Cador, B. Le Guennic and F. Pointillart, Inorg. Chem. Front., 2019, 6, 3398–33417 RSC; (b) Z. Zhu, X.-L. Li, S. Liu and J. Tang, Inorg. Chem. Front., 2020, 7, 3315–3326 RSC; (c) M. Feng, Z.-Y. Ruan, Y.-C. Chen and M.-L. Tong, Chem. Commun., 2020, 56, 13702–13718 RSC.
- (a) M. Irie, T. Fulcaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114(24), 12174–12277 CrossRef CAS PubMed; (b) D. Pinkowicz, M. Ren, L. M. Zheng, S. Sato, M. Hasegawa, M. Morimoto, M. Irie, B. K. Breedlove, G. Cosquer, K. Katoh and M. Yamashita, Chem. – Eur. J., 2014, 20(39), 12502–12513 CrossRef CAS PubMed; (c) K. Rogacz, M. Brzozowska, S. Baś, K. Kurpiewska and D. Pinkowicz, Inorg. Chem., 2022, 61(41), 16295–16306 CrossRef CAS PubMed.
- M. Hojorat, H. Al Sabea, L. Norel, K. Bernot, T. Roisnel, F. Gendron, B. Le Guennic, E. Trzop, E. Collet, J. R. Long and S. Rigaut, J. Am. Chem. Soc., 2019, 142, 931–936 CrossRef PubMed.
- S.-C. Wei, M. Pan, Y.-Z. Fan, H. Liu, J. Zhang and C.-Y. Su, Chem. – Eur. J., 2015, 21, 7418–7427 CrossRef CAS PubMed.
- L. Norel, L. E. Darago, B. Le Guennic, K. Chakarawet, M. I. Gonzalez, J. H. Olshansky, S. Rigaut and J. R. Long, Angew. Chem., Int. Ed., 2018, 57, 1933–1938 CrossRef CAS PubMed.
- A. Ruiz-Martínez, D. Casanova and S. Alvarez, Chem. – Eur. J., 2008, 14, 1291–1303 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2221723, 2239099, 2239100, 2207245 and 2207246. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc01037f |
| This journal is © The Royal Society of Chemistry 2023 |