
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N-Acyl-1,2,3-triazoles – key intermediates in denitrogenative transformations†
Vladimir
Motornov
 *,
Radek
Pohl
,
Blanka
Klepetářová
and
Petr
Beier
*
*,
Radek
Pohl
,
Blanka
Klepetářová
and
Petr
Beier
*
Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Flemingovo nám. 2, Praha 166 10, Czech Republic. E-mail: motornov@uochb.cas.cz; beier@uochb.cas.cz
First published on 29th June 2023
Abstract
Elusive N-acyl-1,2,3-triazoles formed by direct acylation of NH-1,2,3-triazoles were isolated and fully characterized, including X-ray crystallography. A preference for the formation of thermodynamic N2 isomers was established. Direct evidence of interconversion between N1- and N2-acyltriazoles confirmed their value in denitrogenative transformations. Efficient synthesis of enamido triflates from NH-triazoles via the intermediacy of N2-acyl-1,2,3-triazoles was developed.
1,2,3-Triazoles are important heterocyclic compounds with various biological activities and high synthetic value.1 There are two general synthetic routes to N-substituted 1,2,3-triazoles – cycloaddition reactions of azides with alkynes,2 activated ketones,3 or nitroalkenes,4 and nitrogen functionalization of NH-1,2,3-triazoles with electrophiles.5 Whereas the cycloaddition reaction is widely investigated and established as an efficient and robust method, functionalization of NH-1,2,3-triazoles containing three nucleophilic nitrogen atoms is considered problematic because of unpredictable regioselectivity.1a,5 However, NH-1,2,3-triazoles are available starting materials that can be efficiently prepared from inexpensive NaN36 or TMSN37 and alkynes, or NaN3, aldehydes and nitroalkanes via tandem Henry reaction/[3+2] cycloaddition.8 Recently, a number of regioselective protocols for the synthesis of either N29 or N1-substituted10 isomers of 1,2,3-triazoles have been developed. Thus, alkylation, arylation, vinylation and Michael addition with NH-1,2,3-triazoles as nucleophiles have been described and extensively studied.5,9,10 Sulfonylation of NH-1,2,3-triazoles, which would be a tempting route to synthetically useful N1-sulfonyl-1,2,3-triazoles, is not regioselective (Scheme 1B),11 and only regioselective N2-sulfonylation via radical reaction is known.12
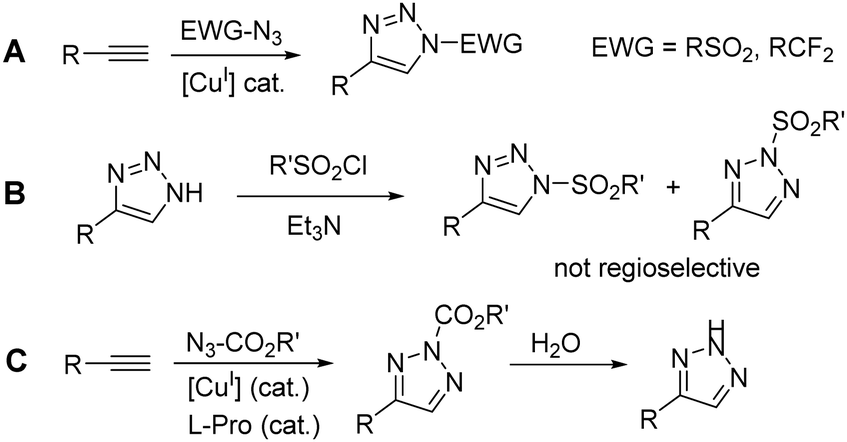 | ||
| Scheme 1 Synthetic approaches to N-EWG-substituted 1,2,3-triazoles. | ||
In contrast to a broad variety of known N-alkyl-, N-fluoroalkyl, N-aryl- and N-sulfonyl-1,2,3-triazoles (Scheme 1A), N-acyl-1,2,3-triazoles are almost unexplored, and only rare and scattered examples have been reported.13 Indeed, a click reaction with acyl azide is not a viable route because of the low stability of acyl azides in the presence of a Cu(I) catalyst, resulting in nitrene formation.14N-Carbamoylation of NH-1,2,3-triazoles was studied giving a mixture of N1- and N2-isomers.15 These compounds possess significant application potential in biological studies. For example, 2-carbamoyl-4-aryl-1,2,3-triazole derivatives were used for site-selective incorporation into proteins,15a as selective chemical probes of endocannabinoid biosynthesis enzymes.15b,c Very recently, the formation of N2-alkoxycarbonyl-1,2,3-triazoles was observed in click reaction of carbamoyl azides with alkynes due to spontaneous carbamoyl group migration to N2-position.16 The resulting N2-carbamoyl triazoles were highly sensitive to hydrolysis to NH-triazoles (Scheme 1C).
1,2,3-Triazoles bearing an electron-withdrawing group at position N1 are useful starting materials in denitrogenative ring opening transformations.1,17 Among them, N-sulfonyl-1,2,3-triazoles are the most widely explored in denitrogenative transformations, which are possible under metal catalysis17 or by the action of Lewis or Brønsted acids (Scheme 2A).18 The transannulation process was recently extended by us to N-fluoroalkylated triazoles.19 Moreover, there was one report about denitrogenative cleavage of N-(1,2,4-triazolyl)-1,2,3-triazoles.20 However, it is remarkable that N-acyltriazoles have never been used as substrates in denitrogenative transformations. This can be attributed to the low stability and propensity to deacylation.16 Also, reactions of N-acylbenzotriazoles with nucleophiles (amines, alcohols) result in acyl group transfer as well.21 Only recently, the first reports of efficient monocyclic 1,2,3-triazole ring cleavage starting from NH-triazoles via in situ acylation were published by us (Scheme 2B)22 and Li's group (Scheme 2C).23 The formation of unstable N-acyltriazoles as intermediates in these processes has been proposed. Indeed, the utilization of readily available NH-triazoles is more atom-economical compared to N-sulfonyl- and N-fluoroalkyl-triazoles. However, the formation, stability and reactivity of N-acyl-1,2,3-triazoles remains unexplored. A high efficiency of NH-triazole ring cleavage transformations could support the hypothesis of regioselective N1-acylation,23 while it is well-established that in reactions of NH-triazoles with electrophiles the formation of N2-substituted isomers is preferred.9h–j We hypothesized that a rapid interconversion between N1 and N2-acyltriazoles in the presence of an excess of acylation agent might enable the conversion of N-acyltriazole into ring cleavage products. Herein we report on our systematic study on N-acylation of 1,2,3-NH-triazoles and their regioselectivity, as well as on our study of ring cleavage reactions of N-acyltriazoles with Brønsted and Lewis acids.
 | ||
| Scheme 2 Transformations of N-EWG-substituted 1,2,3-triazoles and denitrogenative reactions of NH-1,2,3-triazoles with acylating reagents. | ||
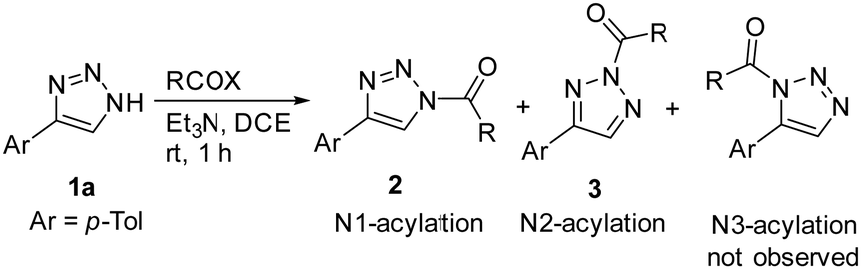
We initiated our study of the acylation of the model 1,2,3-triazole 1a with different acylating agents in the presence of stoichiometric amount of base (Et3N). Acylation of 1a proceeded quickly at ambient temperature, with nearly quantitative yields (Table 1). In the case of benzoyl chloride or benzoic anhydride, mostly N2-acylated triazole 3a formed (entries 1 and 2). The more electron-deficient 4-nitrobenzoyl chloride led to the exclusive formation of N2-substituted acyl triazole 3b (entry 3), while 4-methoxybenzoylchloride, by contrast, showed a preference for N1-isomer 2c (entry 4). The presence of halogen atoms in ortho-positions, exerting steric hindrance, also favoured the formation of N1-isomers (entries 5 and 6). Acetic anhydride and fluorinated acid anhydrides afforded almost exclusively N2-isomers (entries 7–9). However, ethyl chloroformate gave a nearly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of isomers (entry 10). Thus, the general observation is that N2-acylation is favoured and that only soft, weak and bulky acylating reagents favour N1-acylation. Solvent effects and effects of substituents at position 4 affect only slightly the reaction outcome, compared to the remarkable effect of acylating agents (see the ESI† for full details).
:1 mixture of isomers (entry 10). Thus, the general observation is that N2-acylation is favoured and that only soft, weak and bulky acylating reagents favour N1-acylation. Solvent effects and effects of substituents at position 4 affect only slightly the reaction outcome, compared to the remarkable effect of acylating agents (see the ESI† for full details).
|
|
||||
|---|---|---|---|---|
| Entry | RCOX | Products | Yieldb (%) 2 + 3 | Acylation ratioc2/3 |
| a Reaction conditions: 1a (0.10 mmol) and RCOX (0.10-0.11 mmol) mmol), Et3N (0.10-0.11 mmol), DCE (0.5 ml), rt, 1 h. b Isolated yield. c Determined by 1H NMR. d Yield of crude products, not purified from triethylammonium salt. | ||||
| 1 | PhCOCl | 2a + 3a | >98 | 14:86 |
| 2 | (PhCO)2O | 2a + 3a | >98 | 8:92 |
| 3 | 4-O2N–C6H4COCl | 3b | quant.d | <1:99 |
| 4 | 4-MeO–C6H4COCl | 2c + 3c | >98 | 73:27 |
| 5 | 2-Cl–C6H4COCl | 2d + 3d | 94 | 80:20 |
| 6 | 2-Br–C6H4COCl | 2e + 3e | 96 | 79:21 |
| 7 | Ac2O | 2f + 3f | 98 | 5:95 |
| 8 | (HCF2CO)2O | 3g | quant.d | <1:99 |
| 9 | (CF3CO)2O | 3h | quant.d | <1:99 |
| 10 | ClCO2Et | 2i + 3i | 97 | 45:55 |
X-ray diffraction analysis of N1-isomer 2c and N2-isomer 3f confirmed their structure (Fig. 1). Surprisingly, acylated triazoles 2 and 3 are stable enough to be isolated by solvent extraction using aqueous workup and to be fully characterized spectroscopically. Products 3b, 3g and 3h were found to be hydrolytically very unstable and aqueous workup could not be used. All acylated triazoles underwent partial or full decomposition during silica gel column chromatography.
 | ||
| Fig. 1 X-ray crystal structures of 2c (left, CCDC 2244997) and 3f (right, CCDC 2244996). Ellipsoids are set at a 50% probability level. | ||
The structures of the obtained N1- and N2-acyltriazoles were additionally confirmed by 1H-15N HMBC NMR data. 1H-15N HMBC data values, namely δ(15N) and JHN were compared with calculated values for 4-phenyl-N-difluoroacetyl-1,2,3-triazoles, which confirmed the presence of a difluoroacetyl group at the N2 position (see ESI†). Importantly, 1H–15N HMBC NMR of 2c confirmed the presence of an acyl group at position N1; this result is corroborated by single crystal X-ray analysis data (Fig. 1). Regarding 1H NMR of N-acyltriazole mixtures, the isomers could be identified by their chemical shifts of H5, which was shifted by 0.2–0.4 ppm downfield for N1-acyltriazoles (δH5 = 8.4–8.7 ppm) compared to N2-acyltriazoles (δH5 = 8.1–8.3 ppm). This tendency corresponds to literature δH5 shifts observed for N1- and N2-tosyl-4-phenyl-1,2,3-triazoles.12,24
N1- and N2-isomers of acylated triazoles convert to one another. The N1- to N2-acyltriazole interconversion (Scheme 3) can be driven by thermodynamics, as in the case of the slow conversion of 2c to 3c at room temperature, or by the formation of a crystal lattice during crystallization of sterically hindered N2-isomer 3e (Scheme 3). By contrast, we succeeded in isolating pure N1-isomer 2c by recrystallization of a 2c/3c mixture. The process of N1- to N2-interconversion depends on solvent, the concentration and even the reaction scale, which complicated its detailed study.
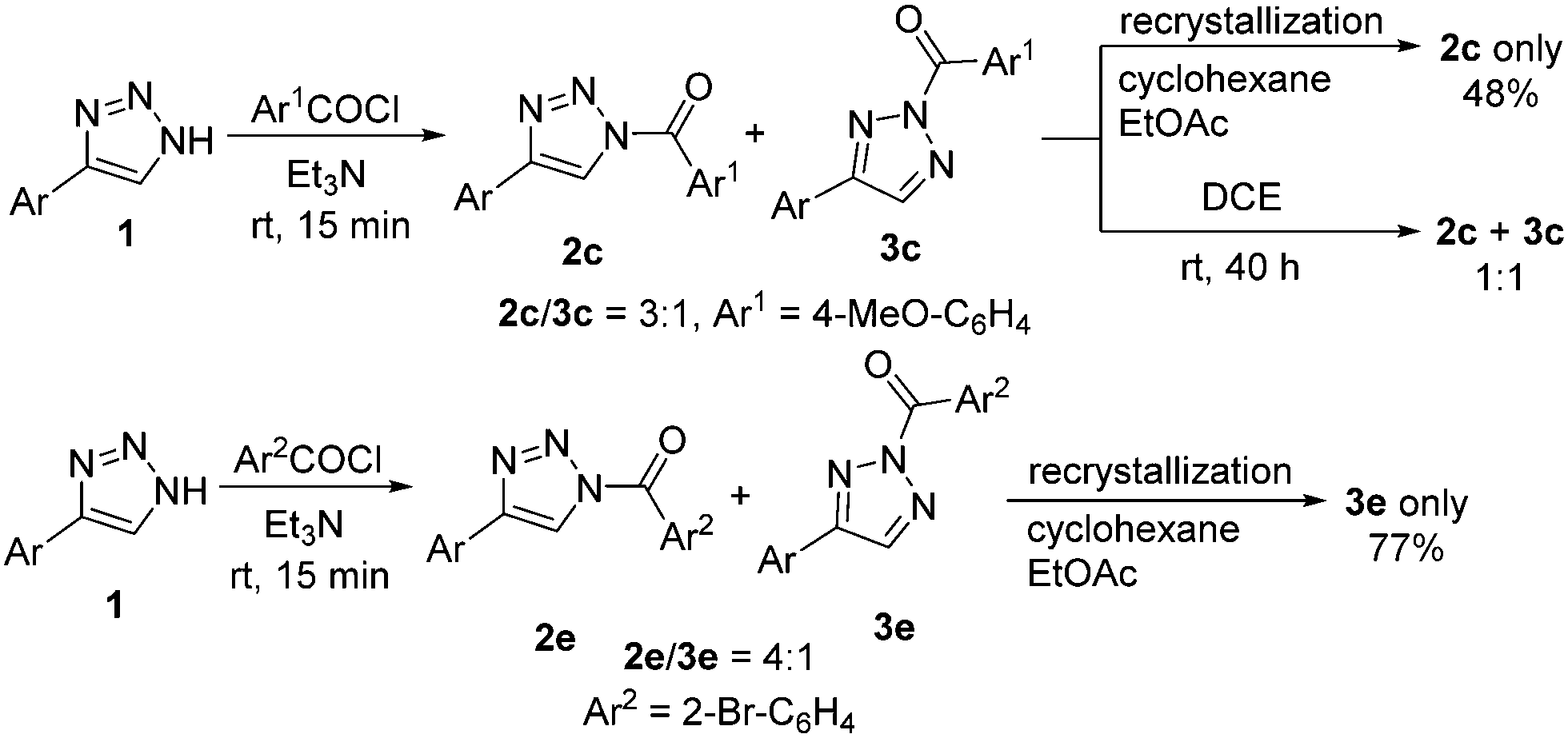 | ||
| Scheme 3 Evidence of N1- to N2-acyltriazole interconversion process (Ar = p-tolyl). | ||
Nevertheless, denitrogenative triazole ring cleavage can only take place from N1 isomers. We demonstrated that the reverse process of N2- to N1-acyltriazole interconversion is crucial for the success of N-acyltriazole cleavage with the involvement of N2-isomers in denitrogenative transformations. In situ formed N2-acylated triazoles 3 react efficiently with triflic acid to produce enamido triflates 4 (Scheme 4). Previously, trifluoroacetylated enamido triflates were prepared by the reaction of N-perfluoroalkyl-1,2,3-triazoles with triflic acid,19e but the present methodology benefits from the use of readily available NH-triazoles and trifluoroacetic anhydride. The reaction was found to be applicable for the synthesis of products 4, bearing neutral aryl substituents (4a, 4b, 4d) or a bromine atom (4c), in good yields. Electron-withdrawing and bulky substituents on the aryl ring were also tolerated (4e–4h). Importantly, the method broadens the scope of enamido triflates to compounds with other fluoroalkyl groups than trifluoromethyl (CF2Cl, CF2CF3, CF2H; 4j–4l), as well as the trichloroacetyl group (4m). 4-Alkyltriazoles giving unstabilized vinyl cation afforded complex mixtures.
 | ||
| Scheme 4 Formation of enamido triflates 4 from NH-triazoles 1. Reaction conditions: 1 (0.3 mmol), (RCO)2O (0.33 mmol), Et3N (0.33 mmol) in DCE (1 ml) stirred for 1 h, then TfOH (0.45–0.54 mmol), 50–80 °C, 3–20 h. | ||
For the above-mentioned transformations, the following mechanism of acyltriazole cleavage to form enamido triflates is proposed: Protonation of acyltriazoles 2 or 3 can theoretically lead to six protonated species, which are in equilibrium due to acyl group and proton shifts. By analogy to our earlier investigations and DFT calculations19f only species A undergoes N1–N2 cleavage to give the diazo/diazonium intermediate B. Diazonium B undergoes denitrogenation to vinyl cation C, and recombination with the triflate anion gives enamido triflate 4 (Scheme 5).
 | ||
| Scheme 5 Mechanism of β-enamido triflate formation. | ||
Acylated triazoles can be also cleaved with Lewis acids. In situ-formed acylated triazoles 2 and 3 reacted with an equimolar amount of AlX3 to produce β-haloenamides 5, presumably via cleavage of forming N1-acyltriazole to a vinyl cation (Scheme 6). The less electron-deficient N1- or N2-acylated triazoles 2c, 3c, or 3e cyclized to give oxazoles 6, showing that both isomers can be converted to the same ring-cleavage and cyclization products.
 | ||
| Scheme 6 Ring cleavage of acylated triazoles using Lewis acids. | ||
In conclusion, N-acylation of 1,2,3-NH-triazoles was investigated and N2-acyltriazoles were found to be the main products; however, electron-rich and bulky acylating reagents induced the formation of a mixture of N1- and N2-acylated triazoles. Interconversion between regioisomers of N-acyltriazoles under thermodynamic conditions, during crystal formation, or in the presence of Brønsted or Lewis acids was observed. Triazole denitrogenative ring cleavage starting from N2-acyltriazoles is reported for the first time. Efficient synthetic access to valuable vinyl triflates from NH-triazoles was developed.
This work was supported by the Czech Academy of Sciences (RVO: 61388963) and the Czech Science Foundation (23-04659S).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Chemistry of 1,2,3-triazoles, ed. W. Dehaen and V. A. Bakulev, Springer, Cham, 2015 Search PubMed; (b) H. M. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151 RSC.
- (a) M. Meldal and F. Diness, Trends Chem., 2020, 2, 569 CrossRef CAS; (b) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302 RSC; (c) L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933 CrossRef CAS; (d) F. Wei, W. Wang, Y. Ma, C.-H. Tung and Z. Hu, Chem. Commun., 2016, 52, 14188 RSC.
- (a) R. Prakash, T. Opsomer and W. Dehaen, Chem. Rec., 2021, 21, 376 CrossRef CAS PubMed; (b) J. John, J. Thomas and W. Dehaen, Chem. Commun., 2015, 51, 10797 RSC.
- (a) C. P. Parambil, S. P. Veethil and W. Dehaen, Synthesis, 2022, 910 CrossRef; (b) V. A. Motornov, S. L. Ioffe and A. A. Tabolin, Targets Heterocycl. Syst., 2019, 23, 237 CAS.
- (a) T. Opsomer and W. Dehaen, Chem. Commun., 2021, 57, 1568 RSC; (b) C. P. Parambil, S. P. Veethil and W. Dehaen, Synthesis, 2022, 910 CrossRef.
- (a) D. Jankovic, M. Virant and M. Gazvoda, J. Org. Chem., 2022, 87, 4018 CrossRef CAS PubMed; (b) L. Hu, C. Muck-Lichtenfield, T. Wang, G. He, M. Gao and J. Zhao, Chem. – Eur. J., 2016, 22, 911 CrossRef CAS PubMed.
- T. Jin, S. Kamijo and Y. Yamamoto, Eur. J. Org. Chem., 2004, 3789 CrossRef CAS.
- (a) R. Hui, M. Zhao, M. Chen, Z. Ren and Z. Guan, Chin. J. Chem., 2017, 35, 1808 CrossRef CAS; (b) L. Wu, X. Wang, Y. Chen, Q. Huang, Q. Lin and M. Wu, Synlett, 2016, 437 CAS.
- (a) For a review, see: Y. Zheng, L. Tian, V. Ramadoss, H. Zhang, L.-L. Zhu and Y. Wang, Synthesis, 2022, 2548 CAS Recent representative examples: ; (b) S. Roshandel, M. J. Lunn, G. Rasul, D. S. M. Ravinson, S. C. Suri and G. K. S. Prakash, Org. Lett., 2019, 21, 6255 CrossRef CAS PubMed; (c) V. Motornov, G. V. Latyshev, Y. N. Kotovschikov, N. V. Lukashev and I. P. Beletskaya, Adv. Synth. Catal., 2019, 361, 3306 CrossRef CAS; (d) X. Man, Y. C. Li, X. X. Li and Z. G. Zhao, New J. Chem., 2019, 43, 14739 RSC; (e) V. A. Motornov, A. A. Tabolin, R. A. Novikov, N. E. Shepel, V. G. Nenajdenko and S. L. Ioffe, Tetrahedron, 2018, 74, 3897 CrossRef CAS; (f) X. Deng, X. Lei, G. Nie, L. Jia, Y. Li and Y. Chen, J. Org. Chem., 2017, 82, 6163 CrossRef CAS PubMed; (g) S. Ueda, M. Su, B. P. Fors and S. L. Buchwald, J. Org. Chem., 2012, 77, 2543 CrossRef CAS PubMed; (h) Y. Chen, Y. Liu, J. L. Petersen and X. Shi, Chem. Commun., 2008, 44, 3254 RSC; (i) Y. Liu, W. Yan, Y. Chen, J. L. Petersen and X. Shi, Org. Lett., 2008, 10, 5389 CrossRef CAS PubMed; (j) X. Wang, L. Zhang, H. Lee, N. Haddad, D. Krishnamurthy and C. H. Senanayake, Org. Lett., 2009, 11, 5026 CrossRef CAS PubMed.
- (a) K. Morimoto, Y. Ohnishi, A. Nakamura, K. Sakamoto, T. Dohi and Y. Kita, Asian J. Org. Chem., 2014, 3, 382 CrossRef CAS; (b) C. Sun, X. Yian, Y. Li, X. Li and Z. Zhao, Org. Biomol. Chem., 2017, 15, 2721 RSC; (c) S. P. Desai and M. S. Taylor, Org. Lett., 2021, 23, 7049 CrossRef CAS PubMed; (d) S. P. Desai, M. T. Zambri and M. S. Taylor, J. Org. Chem., 2022, 87, 5385 CrossRef CAS PubMed.
- T. V. Beryozkina, et al. , Tetrahedron, 2015, 71, 6189 CrossRef CAS.
- R. J. Reddy, A. Shankar, Md Waheed and J. B. Nanubolu, Tetrahedron Lett., 2018, 59, 2014 CrossRef CAS.
- For inconsistent data regarding regioselectivity of triazole acylation, see: (a) Y. Takanashi, et al. , J. Org. Chem., 2017, 82, 11370 CrossRef PubMed; (b) I. S. Odin, S. Cao, D. Hughes, E. V. Zamaratskii, Yu. P. Zarubin, P. P. Purygin, A. A. Golovanov and S. S. Zlotskii, Dokl. Chem., 2020, 492, 89 CrossRef CAS; (c) R. Birkofer and P. Wegner, Chem. Ber., 1967, 100, 3485 CrossRef; (d) F. Politano, A. L. Sandoval, M. L. Witko, K. E. Doherty, C. M. Schroeder and N. E. Leadbeater, Eur. J. Org. Chem., 2022, 38 Search PubMed.
- E. Haldon, M. Besora, I. Cano, X. C. Cambeiro, M. A. Pericas, F. Maseras, M. C. Nicasio and P. J. Perez, Chem. – Eur. J., 2014, 20, 3463 CrossRef CAS PubMed.
- (a) Q. Lin, A. Rahim and Y. Xu, Angew. Chem., Int. Ed., 2022, 61, e202202657 Search PubMed; (b) K. van Rooden, et al. , Chem. – Asian J., 2018, 13, 3491 CrossRef PubMed; (c) K.-L. Hsu, K. Tsuboi, L. R. Whitby, A. E. Speers, H. Pugh, J. Inloes and B. F. Cravatt, J. Med. Chem., 2013, 56, 8257 CrossRef CAS PubMed; (d) L. Chen, L. J. Keller, E. Cordasco, M. Bogyo and C. S. Lentz, Angew. Chem., Int. Ed., 2019, 58, 5643 CrossRef CAS PubMed.
- B. Chakraborti, T. Mandal, C. P. Roy and J. Dash, Chem. Commun., 2021, 57, 7970 RSC.
- (a) M. Akter, K. Rupa and P. Anbarasan, Chem. Rev., 2022, 122, 13108 CrossRef CAS PubMed; (b) B. Chattopadhyay and V. Gevorgyan, Angew. Chem., Int. Ed., 2012, 51, 862 CrossRef CAS PubMed; (c) P. Anbarasan, D. Yadagiri and S. Rajasekar, Synthesis, 2014, 3004 CrossRef CAS.
- (a) Z.-F. Xu, H. C. Dai, L. H. Shan and C.-Y. Li, Org. Lett., 2018, 20, 1054 CrossRef CAS PubMed; (b) D. Yang, L. Shan, Z.-F. Xu and C.-Y. Li, Org. Biomol. Chem., 2018, 16, 1461 RSC.
- (a) V. Motornov, A. Markos and P. Beier, Chem. Commun., 2018, 54, 3258 RSC; (b) V. Motornov and P. Beier, J. Org. Chem., 2018, 83, 15195 CrossRef CAS PubMed; (c) V. Motornov, V. Košťál, A. Markos, D. Taffner and P. Beier, Org. Chem. Front., 2019, 6, 3776 RSC; (d) O. Bakhanovich, V. Khutorianskyi, V. Motornov and P. Beier, Beilstein J. Org. Chem., 2021, 17, 504 CrossRef CAS PubMed; (e) A. Markos, S. Voltrová, V. Motornov, D. Tichý, B. Klepetářová and P. Beier, Chem. – Eur. J, 2019, 25, 7640 CrossRef CAS PubMed; (f) A. Markos, L. Janecký, T. Chvojka, T. Martinek, H. Martinez-Seara, B. Klepetářová and P. Beier, Adv. Synth. Catal., 2021, 363, 3258 CrossRef CAS.
- M. Zibinsky and V. V. Fokin, Org. Lett., 2011, 13, 4870 CrossRef CAS PubMed.
- (a) A. R. Katritzky, H.-Y. He and K. Suzuki, J. Org. Chem., 2000, 65, 8210 CrossRef CAS PubMed; (b) A. R. Katritzky, A. A. A. Abdel-Fattah and M. Wang, J. Org. Chem., 2003, 68, 1443 CrossRef CAS PubMed; (c) A. R. Katritzky, A. A. Shestopalov and K. Suzuki, Synthesis, 2004, 1806 CrossRef CAS.
- (a) V. Motornov and P. Beier, Org. Lett., 2022, 24, 1958 CrossRef CAS PubMed; (b) V. Motornov and P. Beier, New J. Chem., 2022, 46, 14318 RSC.
- T. Wang, Z. Tang, H. Luo, Y. Tian, M. Xu, Q. Lu and B. Li, Org. Lett., 2021, 23, 6293 CrossRef CAS PubMed.
- Q. Xing, C. Zhou, S. Jiang, S. Chen and G.-J. Ding, Org. Biomol. Chem., 2021, 19, 7838 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization and copies of NMR spectra. CCDC 2244996 and 2244997. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc00987d |
| This journal is © The Royal Society of Chemistry 2023 |