
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cu-loaded zeolites enable the selective activation of ethane to ethylene at low temperatures and pressure†
Karoline
Kvande
 a,
Sebastian
Prodinger
a,
Bjørn Gading
Solemsli
a,
Silvia
Bordiga
b,
Elisa
Borfecchia
b,
Unni
Olsbye
a,
Pablo
Beato
c and
Stian
Svelle
*a
a,
Sebastian
Prodinger
a,
Bjørn Gading
Solemsli
a,
Silvia
Bordiga
b,
Elisa
Borfecchia
b,
Unni
Olsbye
a,
Pablo
Beato
c and
Stian
Svelle
*a
aCentre for Materials Science and Nanotechnology (SMN), Department of Chemistry, University of Oslo, 1033 Blindern, 0315, Oslo, Norway. E-mail: stian.svelle@kjemi.uio.no
bDepartment of Chemistry, NIS Center and INSTM Reference Center, University of Turin, via P. Giuria 7, 10125 Turin, Italy
cTopsoe A/S, Haldor Topsøes Alle 1, DK-2800 Kgs, Lyngby, Denmark
First published on 18th April 2023
Abstract
Cu-zeolites are found to activate the C–H bond of ethane already at 150 °C in a cyclic protocol and form ethylene with a high selectivity. Both the zeolite topology and Cu content are found to impact the ethylene yield. Ethylene adsorption studies with FT-IR, demonstrate that oligomerization of ethylene occurs over protonic zeolites, while this reaction does not occur over Cu-zeolites. We postulate that this observation is the origin of the high ethylene selectivity. Based on the experimental results, we propose that the reaction proceeds via the formation of an ethoxy intermediate.
The global demand for gas is expected to rise continuously over the coming years, and the situation is confounded by the recent unrest in Europe.1 Furthermore, finding less energy-intensive pathways for the conversion of the components of natural gas remains a high-priority issue. Many direct reaction pathways for methane have been suggested. One of the more promising is a stepwise conversion route over oxygen-activated Cu-zeolites. These can oxidize methane and, with the assistance of water, produce methanol with a selectivity of around 90%.2 The research on this approach has revealed that after oxygen activation, Cu-oxo moieties are formed as the active sites,3 and upon exposure to methane, a methoxy species forms at these active sites in the framework.4 Since the Cu-sites can activate methane, we found it important to investigate whether these sites also are capable of activating other hydrocarbons, like ethane, by applying the same reaction procedure. Today the industrially applied method for converting ethane is by steam cracking to ethylene at 800–900 °C, requiring large amounts of energy.5 Non-oxidative (EDH) and oxidative (ODH) dehydrogenation of ethane are among the most studied pathways for direct ethane conversion.6 EDH typically requires high temperatures and low pressures to push the equilibrium toward product formation due to the endothermic nature of the reaction. However, high temperatures can lead to less controllable gas phase reactions between the reactants and products and therefore often lead to equilibrium constraints for EDH reactions. ODH however, is exothermic, and can therefore be operated at lower temperatures. The typical range is 300–700 °C, and a variety of supported metals, oxides, and mixed-metal catalysts have been investigated for the reaction6b The main drawback of ODH is that measures need to be taken to limit the product over-oxidation to CO2 and to find safe operational modes due to the highly exothermic nature of the reaction.
Partial oxidation of ethane with H2O2 at 50 °C over Fe and Fe/Cu-loaded zeolites is another interesting pathway that has been suggested by Forde et al.7 The authors show that different reaction pathways occur, depending on the ion-exchanged metal. Cu promotes the formation of ethylene, while Fe promotes acetic acid. It is also interesting to note that the partial oxidation of ethane at low temperatures (∼75 °C) has been shown to be possible over an iron-based MOF. The MOF can form Fe-oxo intermediates upon oxidation in N2O, and these species can be used to activate ethane to form ethanol and acetaldehyde.8
Herein, we explore the conversion of ethane based on a stepwise conversion route, optimized for methane to methanol (MTM) conversion,9 over various Cu-loaded zeolites, namely, MOR, ZSM-5, and SSZ-13. All the samples have been well characterized with techniques like XRD, SEM, elemental analysis, and N2-physisorption, in previous publications.9,10 The relevant elemental composition and the samples corresponding activity in the methane to methanol (MTM) conversion is reported in Table S1 in the ESI.† In this fundamental study, we report a new pathway for ethane conversion over Cu-zeolites, and we explore the structural and compositional requirements for an effective reaction. All experimental methods are reported in the ESI,† Section S1.
Already from our initial results, following the scheme reported in ESI,† Section S1.2, it was evident that a Cu-loaded MOR zeolite, 0.36Cu,H-MOR(11), where 0.36 is the Cu/Al ratio and 11 is the Si/Al ratio, was able to activate the C–H bond in ethane. The main product was ethylene, with ∼95% hydrocarbon selectivity during the steam extraction step (ESI,† Fig. S1(a)) as determined with GC-FID. The only other products detected were butene and ethanol. Small traces of CO2 were also observed qualitatively with the MS (ESI,† Fig. S1(b)). When following the productivity throughout three reaction cycles, the reactivity seems to be quasi-stable, with only a small decrease in ethylene yield for each cycle (Fig. 1).
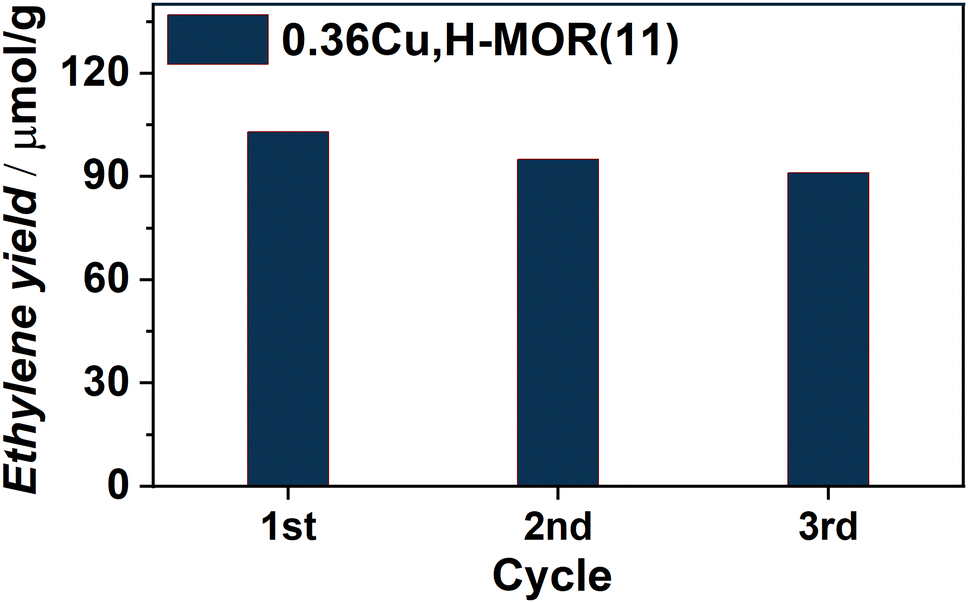 | ||
| Fig. 1 Ethylene yield (μmol g−1) obtained for three consecutive reaction cycles as defined in the ESI,† Scheme S1. | ||
Fig. 2 shows the yield of ethylene obtained over different material topologies and zeolite compositions. Several interesting features of the reaction can be observed. 0.36Cu,H-MOR(11) is used as reference material for the discussion. First, the reaction was performed over three test materials. A Cu-loaded silica (5 wt%Cu–SiO2) containing CuO clusters, a Cu-loaded aluminosilicate with similar Al and Cu content as the reference material that contains [AlO4]− for Cu and H exchange, and an H-MOR zeolite (a porous zeolite framework with acid sites, but no Cu). They all yielded negligible amounts of ethylene and other products, confirming that both well-defined Cu sites and the zeolite framework with restricted exchange sites, are necessary to promote the activation of ethane. Further, a Na-based, Cu-exchanged MOR zeolite (0.32Cu,Na-MOR(11)) with similar Cu-content as the reference material, but less Brønsted sites (see the ESI,† Fig. S2), displayed a similar ethylene yield and product distribution (ESI,† Fig. S1). This implies that the Brønsted sites are not that crucial for ethylene production. This is in contrast to previous findings for the MTM reaction that show that the Brønsted sites play an important part in stabilizing methoxy intermediates for methanol production.4c,11 A Cu-loaded ZSM-5 with similar Cu/Al and Si/Al as our reference material produced significantly lower yield, and thus lower productivity (0.10 molC2H4 molCu−1). This was somewhat surprising, as ZSM-5 has been proposed in other ethane activation reactions, e.g. exchanged with Pt, Zn, Ga, or Fe.7,12 Nevertheless, the same trend between the zeolites has also been observed in the MTM reaction,13 highlighting the similarity of the two reactions. Also a Cu-loaded SSZ-13 was tested in the reaction and proved less productive than both MOR and ZSM-5 (0.07 molC2H4 molCu−1). This evidences a trend, where the productivity increases with increasing pore size: CHA (8r) < MFI (10r) < MOR (12r).
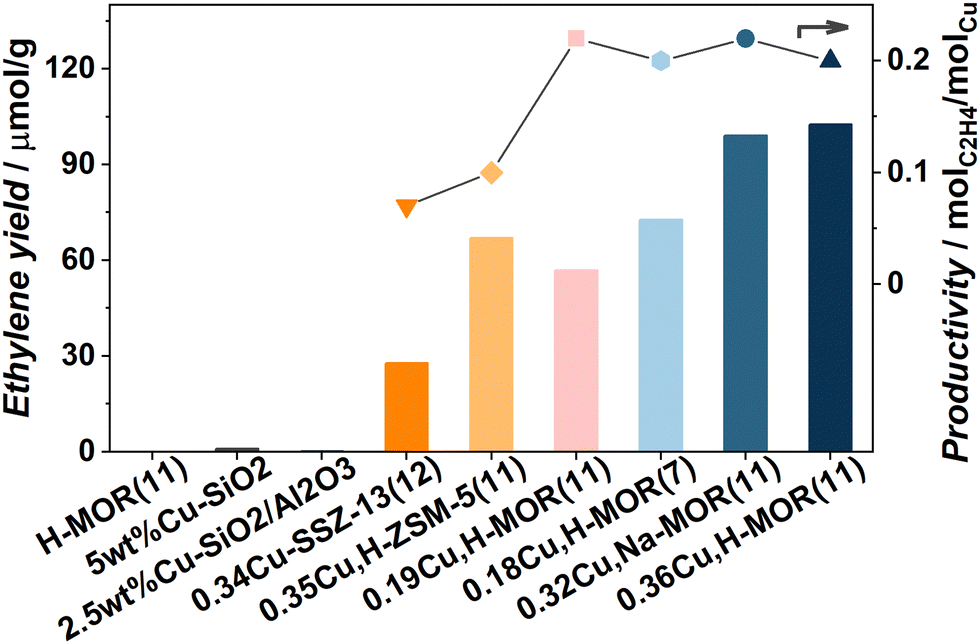 | ||
| Fig. 2 (left axis) Bar plot comparing the ethylene yield (μmol g−1) produced over different materials. The right axis shows the ethylene productivity (molC2H4 molCu−1) for the Cu-zeolites. The line connecting the symbols is for visual guidance. | ||
When employing a Cu-mordenite with both a lower Si/Al and Cu/Al ratio (0.18Cu,H-MOR(7)) that previously has been found to have a remarkably high yield in MTM reaction (ESI,† Table S1), unremarkable results are found when ethane is the reactant. This indicates that the Cu species in 0.18Cu,H-MOR(7) are more efficient at activating methane than ethane, which is surprising given the higher reactivity of ethane (the bond dissociation energies of the first C–H bond in CH4 and C2H6 are 439 vs. 422 kJ mol−1, respectively14). Based on this conspicuous difference, and the lack of a detrimental effect of Na, we conjecture that there might be some subtle differences in the site requirements for methane and ethane activation.
Upon comparing the reference to a sample based on the same zeolite but with lower Cu content (0.19Cu,H-MOR(11)) it is evident that 0.36Cu,H-MOR(11) has the highest yield. The normalized ethylene productivity of the Cu-loaded zeolites (molC2H4 molCu−1) is plotted on the right axis of Fig. 2. The productivity is the same for all four MOR samples (∼0.20 molC2H4 molCu−1), suggesting that regardless of Si/Al ratio or Cu content, the number of active sites increases linearly with the amount of Cu present. In contrast, a volcano-type behavior is typically observed within the same range of Cu content for MTM with the productivity peaking at intermediate Cu-loadings.3a,15 This major difference between the two reactions again indicates different Cu site requirements for C–H activation in ethane vs. methane.
In an attempt to optimize the performance of the reaction, 0.36Cu,H-MOR(11) was exposed to different ethane loading times and temperatures (Fig. 3). First, the results revealed that a somewhat lengthy exposure time was necessary for ethane activation. However, the ethane activation observed herein is much faster than methane activation in the same protocol, as Pappas et al. deemed an exposure time of 6 h necessary to optimize the MTM reaction protocol over Cu-CHA.9 Secondly, upon comparing the ethane loading temperature, a peak productivity of 0.29 molC2H4 molCu−1 was found at 200 °C, before the reactivity started to decrease again at higher temperatures. This behavior is very similar to the MTM reaction.
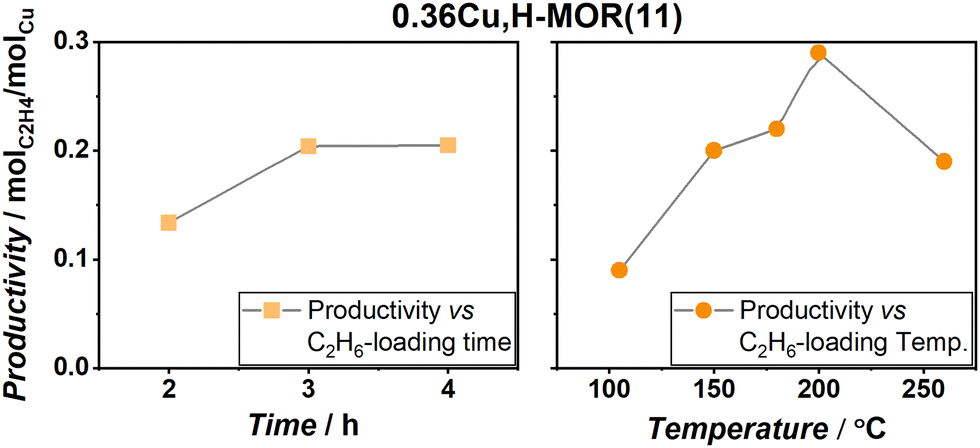 | ||
| Fig. 3 Normalized ethylene productivity (molC2H4 molCu−1) obtained during a systematic study of the reaction conditions. The change in productivity is reported vs. the exposure times (left column – squares) and the temperature (right column – circles). The water extraction step was always kept isothermal to the ethane exposure temperature. The grey lines are only present as a guide for the eye. | ||
To study the interaction of the ethylene product with the materials, 0.36Cu,H-MOR(11) and H-MOR were investigated with FT-IR spectroscopy upon incremental doses of ethylene. The full spectra, including a more detailed description, are reported in ESI† (Section S4, Fig. S3 and S4), while the ν(C–H) region is shown in Fig. 4. Below 3000 cm−1, several new bands evolve for H-MOR(11), but not for the Cu-containing sample. These bands are related to the formation of saturated CH2 (2928 and 2857 cm−1) and CH3 (2960 and 2873 cm−1) groups, indicating that ethylene starts to be polymerized in the H-MOR zeolite. This has also been shown to occur over other H-zeolites.16 Interestingly, this behavior is only observed in the pure, protonic zeolite, and not in the Cu-containing sample, even though 0.36Cu,H-MOR(11) still has a significant amount of Brønsted sites, as seen in the ν(O–H) region of the spectrum before dosing ethylene (ESI,† Fig. S2).
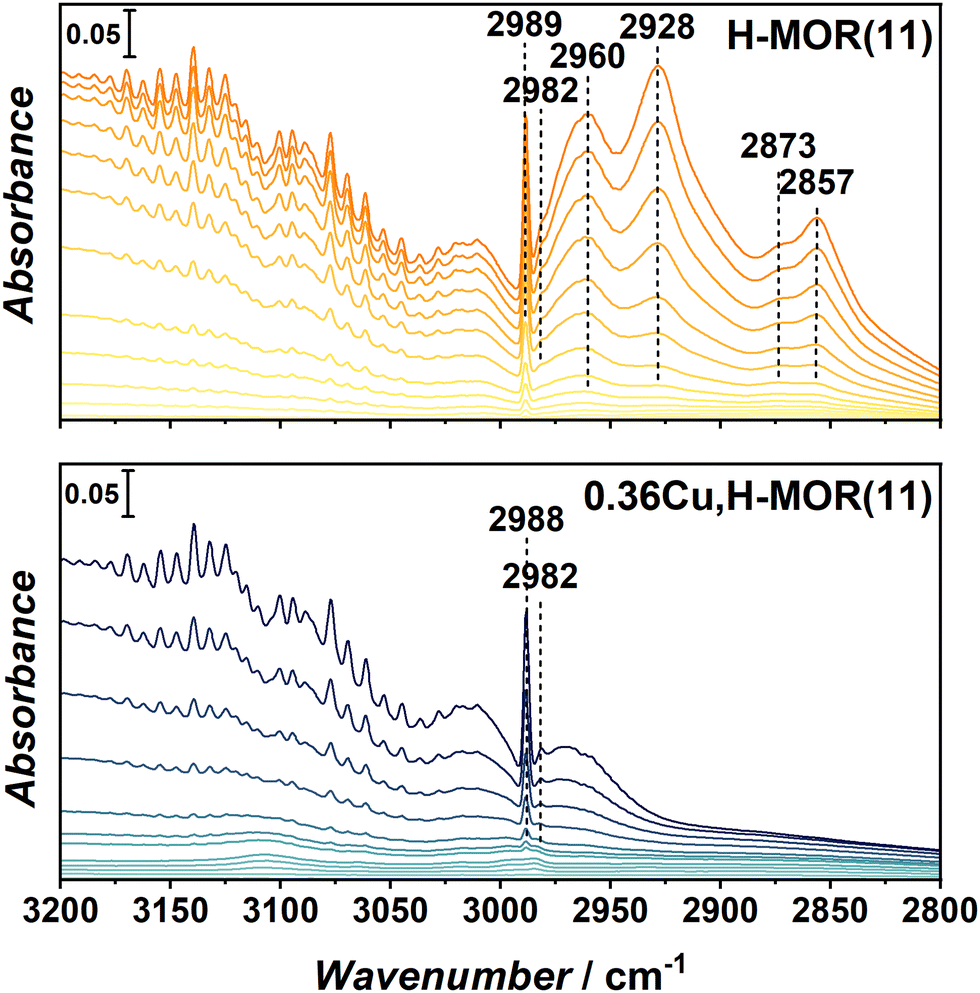 | ||
| Fig. 4 FT-IR spectra of the –CH stretch region obtained for adsorbed ethylene at RT on H-MOR(11) (top panel) and 0.36Cu,H-MOR(11) (bottom panel). The spectra are reported in difference with respect to the spectrum of the activated sample and are normalized to the framework overtones. | ||
To further support that the bands observed for the H-MOR sample indeed originate from oligomerized ethylene, the samples were cooled to 100 K and then outgassed for a longer period up to room temperature (RT). All bands related to oligomerized species that were formed during ethylene dosing remained in the FT-IR spectra after outgassing (ESI,† Fig. S5). We posit that the ability of the Cu-exchanged zeolites to suppress oligomerization might be the key to the high ethylene selectivity. When Cu-zeolites are used in the ETE reaction, the ethylene-derived intermediates are stabilized on the Cu-sites, thus preventing further oxidation or chain growth.
Based on the experimental data obtained, we have tried to identify possible reaction routes for ethylene formation from ethane over activated Cu-sites. The reaction pathways considered are displayed in Scheme 1. No in-depth depiction of the nature of the Cu sites is provided, as this would require advanced characterization and a deeper understanding of the Cu speciation, especially after ethane activation, which goes beyond the scope of this study. There are in principle three pathways for ethylene formation, marked (1)–(3) in Scheme 1. Pathway (1) involves the direct dehydrogenation of ethane and the formation of molecular hydrogen in an endothermic reaction. However, as we have shown that oxidized Cu species are an important factor for ethylene production, and since also C2 oxygenates are observed as (minor) byproducts, it is considerably more likely that the reaction proceeds via an ethoxy intermediate. Accordingly, two exothermic, oxidative reaction pathways, (2) and (3), are proposed. Analogous to methoxy formation in the MTM reaction, ethane could react with some type of CuxOy-moieties leading to ethoxy formation. Pathway (2) is a direct oxidative dehydrogenation of ethane via ethoxy intermediate species. Water facilitates the desorption of ethylene, and H2O (or possibly H2 if the Cu is re-oxidized in this step) is formed as the byproduct. Pathway (3) involves C2 oxygenates (ethanol and possibly diethyl ether (DEE) from dehydration) as free intermediates. Initially formed ethoxy species are detached from the active site upon contact with water. Then, a dehydration process occurs, either instantaneously at the acid sites or some point down the bed, and ethylene is the main product detected in the effluent. To confirm the likelihood of dehydration, we exposed 0.36Cu,H-MOR(11), H-MOR(11), and 0.32Cu,NaMOR(11) directly to ethanol after activation in oxygen, which indeed led to a significant formation of ethylene over all three materials.
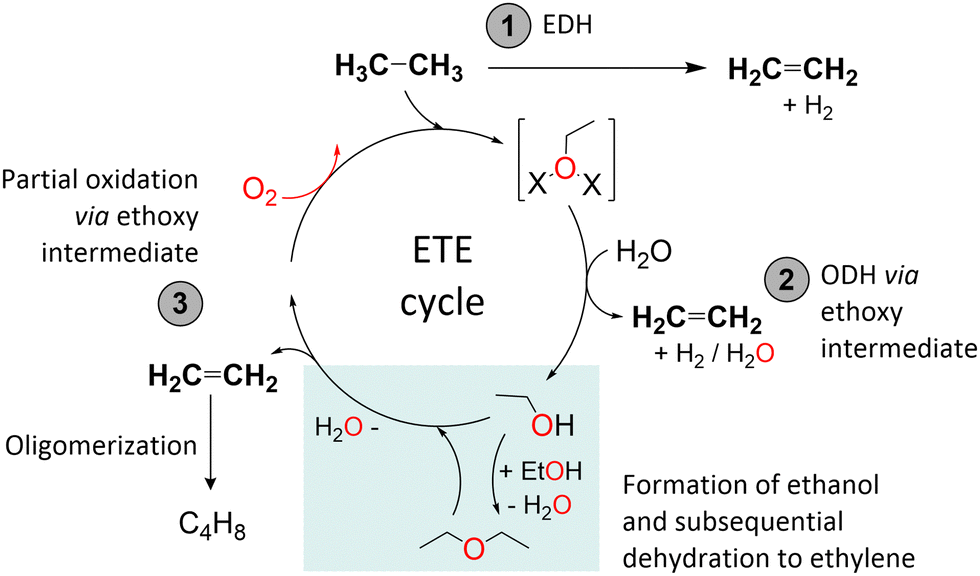 | ||
| Scheme 1 Possible routes derived from the experimental data obtained herein for the ETE reaction. EDH and ODH stand for non-oxidative and oxidative dehydrogenation of ethane, respectively. The colored box highlights the dehydration processes. | ||
Pathway (3) is supported by the observation of trace amounts of ethanol in the effluent. We consider it unlikely that ethanol is formed from acid-catalyzed hydration of ethylene in pathway (2) as the product distribution is the same over both Na-based and H-based Cu-MOR and thus unaffected by the density of Brønsted acid sites (ESI,† Fig. S1). Additionally, since an oxidative dehydrogenation to ethylene, as suggested by pathway (2), should be feasible without the addition of H2O, it is more conceivable that the reaction is proceeding via ethanol formation (pathway (3)). Consequently, the main experimental observations favor reaction pathway (3), and we surmise that the reported ethane activation protocol results in the formation of ethoxy intermediates. We note that this might lay the foundation for a search for a process to form ethanol directly from ethane, by looking for the right reaction conditions and metal-zeolite.
The results reported herein expand the scope of Cu-zeolites in the activation of C–H bonds of lower alkanes. We report, for the first time, a low-temperature, ambient pressure, and highly selective pathway to form ethylene from ethane. The reaction is clearly akin to the well-investigated, stepwise MTM reaction, with about the same temperature needed for C–H activation, but there are also marked differences between these two reactions. Screening of a number of Cu-loaded zeolites indicate that the active site requirements are different for the two substrates. Our work is reconcilable with the formation of an ethoxy intermediate, which might present the opportunity for the selective formation also of ethanol by tuning reaction parameters. As a topic for further studies, we highlight a more detailed investigation that might enable the establishment of a steady-state process, similar to what has been recently described for methane, albeit at very low conversions. This could be an important contribution to the direct C–H activation research.
This publication forms a part of the iCSI (industrial Catalysis Science and Innovation) Centre for Research-based Innovation, which receives financial support from the Research Council of Norway under contract no. 237922. M. Signorile is acknowledged for insightful discussions and G. Deplano for synthesizing the Cu,H-ZSM-5 sample. TOC is created with Biorender.com.
Conflicts of interest
There are no conflicts to declare.References
- Gas Market Report, Q3-2022, IEA, Paris https://www.iea.org/reports/gas-market-report-q3-2022, accessed: 06.10.2022.
- M. A. Newton, A. J. Knorpp, V. L. Sushkevich, D. Palagin and J. A. van Bokhoven, Chem. Soc. Rev., 2020, 49, 1449–1486 RSC
.
-
(a) D. K. Pappas, A. Martini, M. Dyballa, K. Kvande, S. Teketel, K. A. Lomachenko, R. Baran, P. Glatzel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, S. Svelle, P. Beato and E. Borfecchia, J. Am. Chem. Soc., 2018, 140, 15270–15278 CrossRef CAS PubMed
-
(a) E. M. C. Alayon, M. Nachtegaal, A. Bodi and J. A. van Bokhoven, ACS Catal., 2014, 4, 16–22 CrossRef CAS
- T. Ren, M. Patel and K. Blok, Energy, 2006, 31, 425–451 CrossRef CAS
-
(a) H. Saito and Y. Sekine, RSC Adv., 2020, 10, 21427–21453 RSC
- M. M. Forde, R. D. Armstrong, C. Hammond, Q. He, R. L. Jenkins, S. A. Kondrat, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. Willock, C. J. Kiely and G. J. Hutchings, J. Am. Chem. Soc., 2013, 135, 11087–11099 CrossRef CAS PubMed
- D. J. Xiao, E. D. Bloch, J. A. Mason, W. L. Queen, M. R. Hudson, N. Planas, J. Borycz, A. L. Dzubak, P. Verma, K. Lee, F. Bonino, V. Crocellà, J. Yano, S. Bordiga, D. G. Truhlar, L. Gagliardi, C. M. Brown and J. R. Long, Nat. Chem., 2014, 6, 590–595 CrossRef CAS PubMed
- D. K. Pappas, E. Borfecchia, M. Dyballa, I. A. Pankin, K. A. Lomachenko, A. Martini, M. Signorile, S. Teketel, B. Arstad, G. Berlier, C. Lamberti, S. Bordiga, U. Olsbye, K. P. Lillerud, S. Svelle and P. Beato, J. Am. Chem. Soc., 2017, 139, 14961–14975 CrossRef CAS PubMed
-
(a) M. Dyballa, D. K. Pappas, K. Kvande, E. Borfecchia, B. Arstad, P. Beato, U. Olsbye and S. Svelle, ACS Catal., 2019, 9, 365–375 CrossRef CAS
- V. L. Sushkevich, J. A. Van Bokhoven and R. Verel, Angew. Chem., Int. Ed., 2020, 59, 910–918 CrossRef CAS PubMed
-
(a) K.-H. Steinberg, U. Mroczek and F. Roessner, Appl. Catal., 1990, 66, 37–44 CrossRef CAS
- M. B. Park, S. H. Ahn, A. Mansouri, M. Ranocchiari and J. A. van Bokhoven, ChemCatChem, 2017, 9, 3705–3713 CrossRef CAS
- B. Ruscic, J. Phys. Chem. A, 2015, 119, 7810–7837 CrossRef CAS PubMed
- S. Prodinger, K. Kvande, B. Arstad, E. Borfecchia, P. Beato and S. Svelle, ACS Catal., 2022, 12, 2166–2177 CrossRef CAS
- G. Spoto, S. Bordiga, G. Ricchiardi, D. Scarano, A. Zecchina and E. Borello, J. Chem. Soc., Faraday Trans., 1994, 90, 2827–2835 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, physicochemical data, experiment protocol as well as supporting test results and FT-IR spectra (PDF). See DOI: https://doi.org/10.1039/d3cc00948c |
| This journal is © The Royal Society of Chemistry 2023 |