
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDouble-resonance 17O NMR experiments reveal unique configurational information for surface organometallic complexes†
Frédéric A.
Perras
 *ab,
Alejandra
Arroyave
c,
Scott A.
Southern
a,
Jessica V.
Lamb
c,
Yuting
Li
a,
Anne
LaPointe
d and
Massimiliano
Delferro
ce
*ab,
Alejandra
Arroyave
c,
Scott A.
Southern
a,
Jessica V.
Lamb
c,
Yuting
Li
a,
Anne
LaPointe
d and
Massimiliano
Delferro
ce
aChemical and Biological Sciences Division, Ames National laboratory, Ames, IA 50011, USA. E-mail: fperras@ameslab.gov
bDepartment of Chemistry, Iowa State University, Ames, IA 50011, USA
cChemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, Illinois 60439, USA
dDepartment of Chemistry and Chemical Biology, Baker Laboratory, Cornell University, Ithaca, New York 14853, USA
ePritzker School of Molecular Engineering, University of Chicago, Chicago, Illinois 60637, USA
First published on 21st March 2023
Abstract
Obtaining three-dimensional (3D) configurational information of surface organometallic complexes is a persistent challenge due to the low spatial sensitivity of most spectroscopic methods. We show that employing 17O-enriched supports enables highly informative multidimensional NMR experiments, including radial and vertical distance measurements, that can be used to elucidate site geometry.
Surface organometallic chemistry (SOMC) is a popular approach used for the preparation of heterogeneous catalysts with active site structures that are controlled and tunable at the molecular level.1 SOMC catalysts are generally prepared through the protonolysis of a bond on the complex, often a M–X bond, by a surface hydroxyl, leading to the formation of a new covalent interaction between the complex and the support surface. While the ultimate goal is synthesizing sites with well-defined and uniform structures that will have identical reactivities, we and others have underlined that the chemistry is far more complex than is often implied, sometimes leading to many supported species.2–10 Of course, understanding the reactivity of these sites requires knowledge of their structure, however, the most typical characterization tools, such as 1D nuclear magnetic resonance (NMR), infrared spectroscopy, and X-ray absorption spectroscopy (XAS), are insensitive to subtle changes in configuration.
Recent years have seen what is arguably the greatest leap in our understanding of SOMC catalyst structure with the development and generalization of dynamic nuclear polarization (DNP)-enhanced NMR distance measurements.8,10–13 Either alone, or in combination with XAS, these methods have been used to elucidate the first three-dimensional (3D) configurational structures of SOMC complexes. We, for instance, showed that 13C{27Al} distance measurements in an Al2O3-supported Ir pincer complex could be used to distinguish between a dozen different potential grafting configurations,12 and that 13C{29Si} distance measurements in a 29SiO2-supported Sc amidinate complex were able to differentiate different coordination geometries.12
All prior work in this area has mainly been centered on measuring distances involving ligand atoms (13C, 15N, 29Si) while it would be preferable to also involve the metal center. For example, if a ligand is released due to complex decomposition or from protonolysis and remains adsorbed on the surface, it can be challenging to differentiate it from the site of interest. Unfortunately, structural investigations of the metal center are limited to XAS studies in all but a handful of metals (Sc, V, Mn, Co, Y, Ag, Pt), due to most transition metals possessing poor NMR properties.14–20 In this contribution, we show some of the unique opportunities that are made available when SOMC complexes are grafted onto 17O-enriched oxide support materials, specifically silica. Distances can be measured from the nuclei of interest to the 17O-enriched support surface, enabling 3D structure determination. Direct detection of 17O also enables the observation of metal-coordinated oxygen atoms. If the metal center cannot be detected using NMR, these 17O signals can be used as proxies, enabling radial correlations from the center of the complex.
Silica is readily surface-17O-enriched by performing repeated dehydroxylations at an elevated temperature (700 °C) under dynamic vacuum, interspliced with rehydroxylation steps at room temperature with liquid 17O-water (see Fig. 1).21 This procedure is general and can be applied to any oxide support.22–26 At most, this procedure can lead to an enrichment at half the enrichment level of the water at each step, but in practice, we observe roughly a quarter (Fig. 1b). Given that the RESPDOR dephasing rate scales with the spin quantum number of the recoupled nuclide (S = 5/2 for 17O and 1/2 for 29Si), a 40% 17O-enrichment level is predicted to yield roughly equivalent X{17O} RESPDOR dephasing as the corresponding X{29Si} REDOR experiment in a 100% 29Si-enriched silica support (see simulations in Fig. S2, ESI†),27 thus enabling the use of lower enrichment levels. Rotational-echo saturation-pulse double-resonance (RESPDOR)28,29 and rotational-echo double-resonance (REDOR)30 are the most typical heteronuclear distance measurement methods used in the solid-state NMR of quadrupolar and spin-1/2 nuclei, respectively. We found that we could reach over a 60% 17O-enrichment of the surface after four de/rehydroxylation cycles using 90%-enriched 17O-water. 17O NMR spectra of the produced materials revealed that both silanols and siloxane sites are enriched by the procedure.
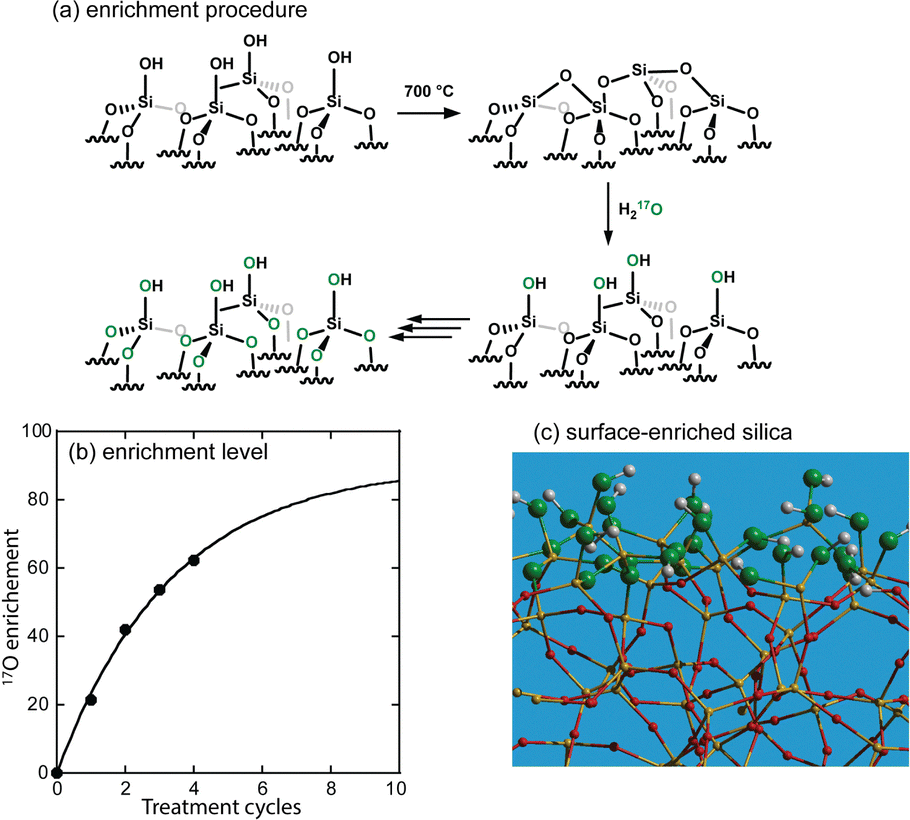 | ||
| Fig. 1 (a) Scheme depicting the stepped 17O enrichment strategy. This strategy leads to the selective enrichment of the topmost layer of the silica, as shown in (c). Progress in the enrichment as a function of de/rehydroxylation cycles is shown in (b). | ||
We recently determined the conformation of a 29SiO2-supported Sc amidinate complex (Fig. 2a) using 13C{29Si} and 1H,13C,15N{45Sc} RE(SP)DOR experiments.12,18 The majority of the configurational information is obtained from the surface-to-atom distance measurements to 29Si, but intramolecular distance measurements were also crucial in confirming the basic molecular structure of the complex. The preparation of 29Si-enriched materials is, however, more expensive and synthetically demanding than enrichment with 17O. We thus grafted the same complex onto a silica gel that we had surface-enriched to roughly 80% with 17O and sought to replicate these earlier results. To this end, we performed DNP-enhanced 13C{17O} phase modulated (PM)-RESPDOR experiments31,32 (Fig. 2b). Coherence lifetimes were somewhat shorter than those obtained using 13C{29Si} REDOR, largely due to the requirement of applying the recoupling pulses to the 13C nuclei. This is counterbalanced, however, by the 2-fold faster dephasing from the stronger dipolar interactions to the spin-5/2 17O nuclei. As such, we were able to acquire comparable data and use this, in conjunction with the INTERFACES program,13 to solve a 3D structure for this complex. The resulting structure is depicted in Fig. 2c, showing a well-defined square pyramidal complex in agreement with what was determined prior.12 The probability ellipsoids are larger for the methyl carbon positions indicating that their positions are less well defined. An overlay of this structure with the best-fit structure determined using 29Si is shown in Fig. 2d, where we can see that the agreement between the two structures is striking, conclusively demonstrating that 17O is a drop-in replacement for 29Si or other nuclei for the determination of the 3D structures of supported sites on oxide supports. Importantly, unlike 29Si, the application of 17O nuclei will remain applicable to other oxides, such as TiO2 and ZrO2 that have as of yet remained out of reach of this type of inquiry.
 | ||
| Fig. 2 (a) Structure of the grafted Sc amidinate complex and (b) its 13C{17O} PM-RESPDOR NMR data. Resonances are assigned per the structure in (a). Fits are depicted as a shaded range representing all structures within a 90% confidence level, as determined using χ2 analysis. The 3D structure determined using the data is depicted in (c) with ORTEP-like probability ellipsoids and is also overlaid in (d) with the structure determined for the same complex supported onto 29SiO2 using 13C{29Si} REDOR. | ||
While it is possible in this example to perform correlations or distance measurements to 45Sc, as we have shown,18 this is generally the exception and not the rule. The vast majority of transition metals are not amenable to direct NMR investigation due to their unfavorable NMR properties. The use of 17O-enriched supports, however, does generate new opportunities for studying the metal centers in the form of metal-induced shifts. As has been demonstrated by Merle et al.,2117O sites coordinating to metal centers from SOMC catalysts are readily identifiable from their shifted 17O NMR signals. The detection of these sites is useful for confirming the complexation of the metal to the support and can also be used, in conjunction with density functional theory (DFT) calculations, to gain some information about the structural and electronic properties of the metal, such as oxidation state. But these signals can also be used as proxies for the metal centers with which they are bonded in more sophisticated and information-rich NMR experiments.33,34 For example, in Fig. 3 we show a 17O{1H} RINEPT-SR412(tt)-QCPMG HETCOR35–37 spectrum acquired on the Sc complex. All three 1H sites are easily distinguished in the 1H dimension and are seen to correlate with the siloxane sites from the support with relative intensities that are roughly equal to their abundances in the complex. Unlike these correlations, however, we see that the metal-bound 17O site shows its strongest correlation to the least abundant 1H site situated closest to the metal, with the second strongest correlation being the second closest, iso-propyl signal. No correlation to the highly abundant methyl 1H sites is detected. This result thus provides further distance constraints for structure determinations and confirms the bidentate coordination of the ligands to the Sc, and the overall molecular structure of the complex. Previous titration experiments have suggested that residual silanol concentrations are low;18 however, we cannot conclusively rule out correlations to silanol protons as these would overlap with 1H resonance C.
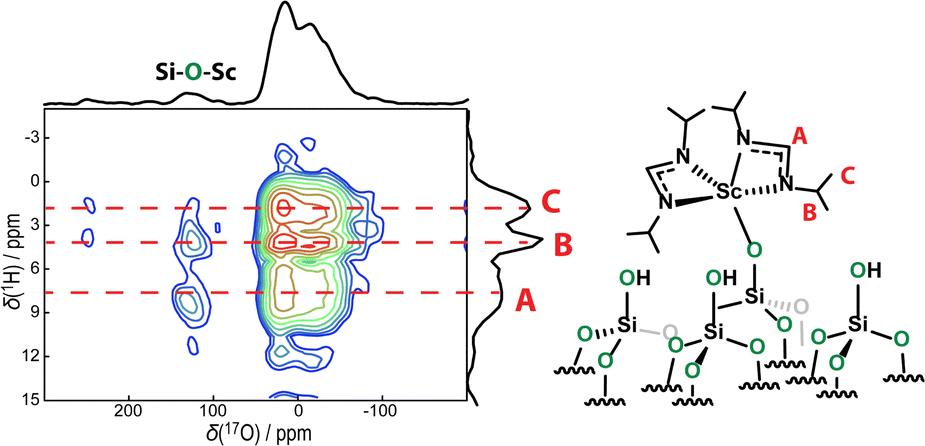 | ||
| Fig. 3 17O{1H} RINEPT-SR412(tt)-QCPMG HETCOR spectrum acquired on the Sc amidinate complex. Correlations show that while correlations between siloxanes and 1H sites on the complex are statistical, those for the metal-bound signal reflect the sites closest to the metal. | ||
We next studied an Ir(POCOP) ethylene complex (POCOP-H: 1,3,5-[(tBu2)PO]3C6H3) that has been used as a heterogeneous dehydrogenation catalyst (Fig. 4).38,39 The complex was grafted onto the same 17O-enriched silica. Its grafting chemistry is, however, more complex than most catalysts, and it has been shown to be able to graft through both the Ir center and the para substituent of the aromatic ring of the POCOP ligand (Fig. 4).39 Distinguishing these two using the 1D NMR spectroscopy of the ligands is challenging as the only possible handle would be chemical shift effects from the Ir-C or Ir-P sites resulting from electronic structure changes, such as oxidation of the metal from Ir(I) to Ir(III).40 Performing these experiments is complicated by the fact that some complexes are degraded following their reaction with the silica, and result in the formation of free di(tert-butyl)phosphine (see ESI†).39
 | ||
| Fig. 4 Potential reaction pathways in the grafting of Ir(POCOP) onto SiO2. | ||
Studying this chemistry is, however, much clearer when utilizing 17O. The DNP-enhanced 1D 17O{1H} RINEPT-SR412(tt)-QCPMG NMR spectrum of the catalyst is shown in Fig. 5b. As can be seen, the only identifiable resonance is that from siloxane species, and we do not detect any Si–O–Ir signals. Using Gauge-including projected augmented wave (GIPAW)41,42 DFT calculations, we predict that such a site would resonate at a center of mass frequency of around 37 ppm on our spectrometer while a Si–O–C site would appear at −116 ppm and be obscured by the solixane resonance. For the DFT investigations, we grafted the complex in both orientations onto a periodic silica amorphous model designed by Ugliengo et al (see ESI†).43,44
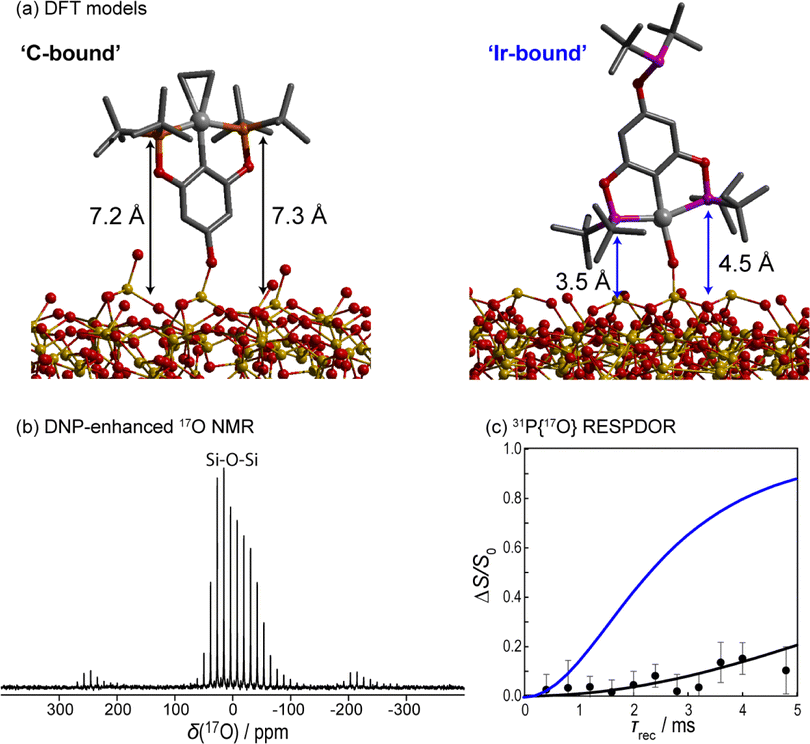 | ||
| Fig. 5 (a) PAW DFT-optimized configurations of both structures from Fig. 4. (b) DNP-enhanced 17O{1H} RINEPT-SR412(tt)-QCPMG NMR spectrum of the material in (a) showing a lack of metal-bound silanols. (c) 31P{17O} PM-RESPDOR results (points) along with fits using the two DFT models shown in (a); Ir-bound in blue and C-bound in black. | ||
Aside from providing an estimate for the chemical shifts, these calculations also enable us to predict the surface-to-atom distances of various sites in the two models. For instance, the two phosphorus atoms are expected to be elevated from the support by 7.2 and 7.3 Å in the carbon-bound model and 3.5 and 4.5 Å in the Ir-bound model. These predictions can be compared directly against an experimental DNP-enhanced 31P{17O} PM-RESPDOR experiment (Fig. 5c), showing clear agreement with the C-bound model. We were unsuccessful in performing 13C{17O} experiments due to the complicated 13C spectrum of the complex (see ESI†).
To conclude, we have shown that there are considerable advantages to 17O-enriching oxide supports used in synthesizing SOMC catalysts. The oxygen spins can be used as a surface probe with which surface-to-atom distances can be measured to determine a complex's 3D conformation or configuration. The metal-bound oxygen center can also be used as a useful proxy for the metal itself and utilized in radial correlations or distance measurements. These types of measurements provide key structural constraints that are unavailable from conventional characterization approaches that focus on molecular structure rather than configuration. This was highlighted by the study of an Ir(POCOP) complex, which we conclusively showed coordinates to the support through its POCOP ligand, rather than through the metal.
This work was supported by the U.S. Department of Energy (DOE), Office of Basic Energy Sciences (BES), Division of Chemical Sciences, Geosciences, and Biosciences (CSGB), as a part of the Ames Laboratory catalysis program, under contracts DE-AC-02-07CH11358 (Ames National Laboratory) and DE-AC-02-06CH11357 (Argonne National Laboratory). The studies of Ir(POCOP) catalyst was supported as part of the Institute for Cooperative Upcycling of Plastics (iCOUP), an Energy Frontier Research Center funded by DOE, BES. Ames Laboratory is operated for the DOE by Iowa State University under contract no. DE-AC02-07CH11358.
Conflicts of interest
There are no conflicts to declare.References
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323 CrossRef PubMed.
- J. Corker, F. Lefebvre, C. Lécuyer, F. Dufaud, F. Quignard, A. Choplin, J. Evans and J.-M. Basset, Science, 1996, 271, 966 CrossRef CAS.
- C. Thieuleux, E. A. Quadrelli, J.-M. Basset, J. Döbler and J. Sauer, Chem. Commun., 2004, 1729 RSC.
- T.-C. Ong, W.-C. Liao, V. Mougel, D. Gajan, A. Ledage, L. Emsley and C. Copéret, Angew. Chem., Int. Ed., 2016, 55, 4743 CrossRef CAS PubMed.
- T. Kobayashi, F. A. Perras, T. W. Goh, T. L. Metz, W. Huang and M. Pruski, J. Phys. Chem. Lett., 2016, 7, 2322 CrossRef CAS PubMed.
- N. Eedugurala, Z. Wang, K. Yan, K. C. Boteju, U. Chaudhary, T. Kobayashi, A. Ellern, I. I. Slowing, M. Pruski and A. D. Sadow, Organometallics, 2017, 36, 1142 CrossRef CAS.
- Z. Wang, S. Patnaik, N. Eedurugala, J. S. Manzano, I. I. Slowing, T. Kobayashi, A. D. Sadow and M. Pruski, J. Am. Chem. Soc., 2020, 142, 2935 CrossRef CAS PubMed.
- F. A. Perras, A. L. Paterson, Z. H. Syed, A. J. Kropf, D. M. Kaphan, M. Delferro and M. Pruski, J. Phys. Chem. C, 2021, 125, 13433 CrossRef CAS.
- Y. Li, U. Kanbur, J. Cui, G. Wang, T. Kobayashi, A. D. Sadow and L. Qi, Angew. Chem., Int. Ed., 2022, 134, e202117394 Search PubMed.
- R. Jabbour, M. Renom-Carrasco, K. W. Chan, L. Völker, P. Berruyer, Z. Wang, C. M. Widdifield, M. Lelli, D. Gajan, C. Copéret, C. Thieuleux and A. Lesage, J. Am. Chem. Soc., 2022, 144, 10270 CrossRef CAS PubMed.
- P. Berruyer, M. Lelli, M. P. Conley, D. L. Silverio, C. K. Widdifield, G. Siddiqi, D. Gajan, A. Lesage, C. Copéret and L. Emsley, J. Am. Chem. Soc., 2017, 139, 849 CrossRef CAS PubMed.
- F. A. Perras, U. Kanbur, A. L. Paterson, P. Chatterjee, I. I. Slowing and A. D. Sadow, Inorg. Chem., 2022, 61, 1067 CrossRef CAS PubMed.
- J. Cunningham and F. A. Perras, J. Magn. Reson. Open, 2022, 11–12, 100066 CrossRef.
- T. Vancompernolle, X. Trivelli, L. Delevoye, F. Pourpoint and R. M. Gauvin, Dalton Trans., 2017, 46, 13176 RSC.
- M. F. Delley, G. Lapadula, F. Núñez-Zarur, A. Comas-Vives, V. Kalendra, G. Jeschke, D. Baabe, M. D. Walter, A. J. Rossini, A. Lesage, L. Emsley, O. Maury and C. Copéret, J. Am. Chem. Soc., 2017, 139, 8855 CrossRef CAS PubMed.
- W. Huynh, D. B. Culver, H. Tafazolian and M. P. Conley, Dalton Trans., 2018, 47, 13063 RSC.
- D. B. Culver, W. Huynh, H. Tafazolian and M. P. Conley, Organometallics, 2020, 39, 1112 CrossRef CAS.
- A. L. Paterson, D.-J. Liu, U. Kanbur, A. D. Sadow and F. A. Perras, Inorg. Chem. Front., 2021, 8, 1416 RSC.
- A. Venkatesh, D. Gioffrè, B. A. Atterberry, L. Rochlitz, S. L. Carnahan, Z. Wang, G. Mendzildjian, A. Lesage, C. Copéret and A. J. Rossini, J. Am. Chem. Soc., 2022, 144, 13511 CrossRef CAS PubMed.
- Z. Wang, L. A. Völker, T. C. Robinson, N. Kaeffer, G. Mendzildjian, R. Jabbour, A. Venkatesh, D. Gajan, A. J. Rossini, C. Copéret and A. Lesage, J. Am. Chem. Soc., 2022, 144, 21530 CrossRef CAS PubMed.
- N. Merle, J. Trébosc, A. Baudouin, I. Del Rosal, L. Maron, K. Szeto, M. Genelot, A. Mortreux, M. Taoufik, L. Delevoye and R. M. Gauvin, J. Am. Chem. Soc., 2012, 134, 9263 CrossRef CAS PubMed.
- M. Wang, X.-P. Wu, S. Zheng, L. Zhao, L. Li, L. Shen, Y. Gao, N. Xue, X. Guo, W. Huang, Z. Gan, F. Blanc, Z. Yu, X. Ke, W. Ding, X.-Q. Gong, C. P. Grey and L. Peng, Sci. Adv., 2015, 1, e1400133 CrossRef PubMed.
- F. A. Perras, Z. Wang, P. Naik, I. I. Slowing and M. Pruski, Angew. Chem., Int. Ed., 2017, 56, 9165 CrossRef CAS PubMed.
- L. Shen, X.-P. Wu, Y. Wang, M. Wang, J. Chen, Y. Li, H. Huo, W. Hou, W. Ding, X.-Q. Gong and L. Peng, J. Phys. Chem. C, 2019, 123, 4158 CrossRef CAS.
- M. Xu, J. Chen, Y. Wen, J.-H. Du, Z. Lin and L. Peng, ACS Omega, 2020, 5, 8355 CrossRef CAS PubMed.
- J. Chen, X.-P. Wu, M. A. Hope, Z. Lin, L. Zhu, Y. Wen, Y. Zhang, T. Qin, J. Wang, T. Liu, X. Xia, D. Wu, X.-Q. Gong, W. Tang, W. Ding, X. Liu, L. Chen, C. P. Grey and L. Peng, Chem. Sci., 2022, 13, 11083 RSC.
- X. Lu, O. Lafon, J. Trébosc and J.-P. Amoureux, J. Magn. Reson., 2012, 215, 34–49 CrossRef CAS PubMed.
- Z. Gan, Chem. Commun., 2006, 4712 RSC.
- L. Chen, X. Lu, Q. Wang, O. Lafon, J. Trébosc, F. Deng and J.-P. Amoureux, J. Magn. Reson., 2010, 206, 269 CrossRef CAS PubMed.
- T. Gullion and J. Schaeffer, J. Magn. Reson., 1989, 81, 196 CAS.
- E. Nimerovski, R. Gupta, J. Yehl, M. Li, T. Polenova and A. Goldbourt, J. Magn. Reson., 2014, 204, 107–113 CrossRef PubMed.
- E. Nimerovski, M. Makrinich and A. Goldbourt, J. Chem. Phys., 2017, 146, 124202 CrossRef PubMed.
- F. A. Perras, K. C. Boteju, I. I. Slowing, A. D. Sadow and M. Pruski, Chem. Commun., 2018, 54, 3472 RSC.
- U. Kanbur, G. Zang, A. L. Paterson, P. Chatterjee, R. A. Hackler, M. Delferro, I. I. Slowing, F. A. Perras, P. Sun and A. D. Sadow, Chem, 2021, 7, 1347 CAS.
- H. Nagashima, A. S. L. Thankamony, J. Trébosc, L. Montagne, G. Kerven, J.-P. Amoureux and O. Lafon, Solid State Nucl. Magn. Reson., 2018, 94, 7 CrossRef CAS PubMed.
- H. Nagashima, J. Trébosc, Y. Kon, K. Sato, O. Lafon and J.-P. Amoureux, J. Am. Chem. Soc., 2020, 142, 10659 CrossRef CAS PubMed.
- H. Nagashima, J. Trébosc, Y. Kon, O. Lafon and J.-P. Amoureux, Magn. Reson. Chem., 2021, 59, 920 CrossRef CAS PubMed.
- Z. Huang, M. Brookhart, A. S. Goldman, S. Kundu, A. Ray, S. L. Scott and B. C. Vicente, Adv. Synth. Catal., 2009, 351, 188 CrossRef CAS.
- B. Shedulko, M. T. Cunningham, A. S. Goldman and F. E. Celik, ACS Catal., 2018, 8, 7828 CrossRef.
- D. M. Kaphan, R. C. Klet, F. A. Perras, M. Pruski, C. Yang, A. J. Kropf and M. Delferro, ACS Catal., 2018, 8, 5363 CrossRef CAS.
- M. Profeta, F. Mauri and C. J. Pickard, J. Am. Chem. Soc., 2003, 125, 541 CrossRef CAS PubMed.
- T. Charpentier, Solid State Nucl. Magn. Reson., 2011, 40, 1 CrossRef CAS PubMed.
- P. Ugliengo, M. Sodupe, F. Musso, I. J. Bush, R. Orlando and R. Dovesi, Adv. Mater., 2008, 20, 4579 CrossRef CAS.
- M. Signorile, C. Salvini, L. Zamirri, F. Bonino, G. Martra, M. Sodupe and P. Ugliendo, Life, 2018, 8, 42 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc00899a |
| This journal is © The Royal Society of Chemistry 2023 |