
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoaxial assembly of helical aromatic foldamers by metal coordination†
Albano
Galan
a,
Kristijan
Lulic
b,
Jingqi
Wang
b,
Barbara
Wicher
 d,
Ivan
Huc
ac,
Jean
Duhamel
b and
Victor
Maurizot
*a
d,
Ivan
Huc
ac,
Jean
Duhamel
b and
Victor
Maurizot
*a
aUniversité de Bordeaux, CNRS, Bordeaux Institut National Polytechnique, CBMN (UMR 5248), Institut Européen de Chimie Biologie, 2 Rue Escarpit, Pessac 33600, France. E-mail: victor.maurizot@u-bordeaux.fr
bInstitute for Polymer Research, Waterloo Institute for Nanotechnology, Department of Chemistry, University of Waterloo, 200 University Avenue West, Waterloo, ON N2L 3G1, Canada. E-mail: jduhamel@uwaterloo.ca
cDepartment Pharmazie, Ludwig-Maximilians-Universität München, Butenandtstraße 5–13, Munich D-81377, Germany
dDepartment of Chemical Technology of Drugs, Poznan University of Medical Sciences, Grunwaldzka 6, 60-780 Poznan, Poland
First published on 3rd April 2023
Abstract
Deprotonation of acid-terminated helical aromatic foldamers with a mineral base in chlorinated solvents led to their dimerization through the coordination of a metal cation (Li+, Na+, K+, Ag+, or Hg2+) with the terminal carboxylate functions. This new ligation method was applied to oligomerize diacid-functionalized foldamers.
Foldamers are artificial oligomers that fold into compact molecular architectures inspired by the secondary structures of biomolecules.1,2 Among the various oligomers and backbones developed in this field, aromatic oligoamides foldamers give access to predictable and robust folded helical conformations with potential applications for protein recognition, as molecular capsules or sensors, or in material science.3–8 Foldamers have also been used as rigid building blocks for the elaboration of more sophisticated self-assembled structures comprised of multiple independent helices.9,10 Among these, an interesting architecture is the piling up of helices into rigid coaxial stacks.11 Such stacks have been promoted in the solid state for example by means of terminal hydrogen-bonding groups12,13 or metal coordination14 as well as in solution,15–18 where they may lead to the formation of fibers.19,20 Here, we introduce a new, simple and robust end-to-end ligation of helical foldamers based on metal coordination in organic solvent to generate well-defined discrete assemblies and supramolecular metallopolymers.
The starting point of this report is a serendipitous finding made during the solution phase step-wise synthesis of 8-amino-2-quinolinecarboxylic acid helical oligoamides (Fig. 1).21 Thus, the incomplete protonation of the carboxylate obtained by saponification of a methyl ester led to the observation of two sets of signals in 1H NMR spectra that were assigned to the sodium carboxylate salt and to the expected acid, respectively. Coordination of metal ions by carboxylates is well known.22–24 Yet, the significant chemical shift differences between the carboxylate and the acid signals and the slow exchange kinetics between the two states on the NMR time scale led us to speculate that the alkaline metal promoted the assembly of oligomers by coordination of their carboxylate end groups.
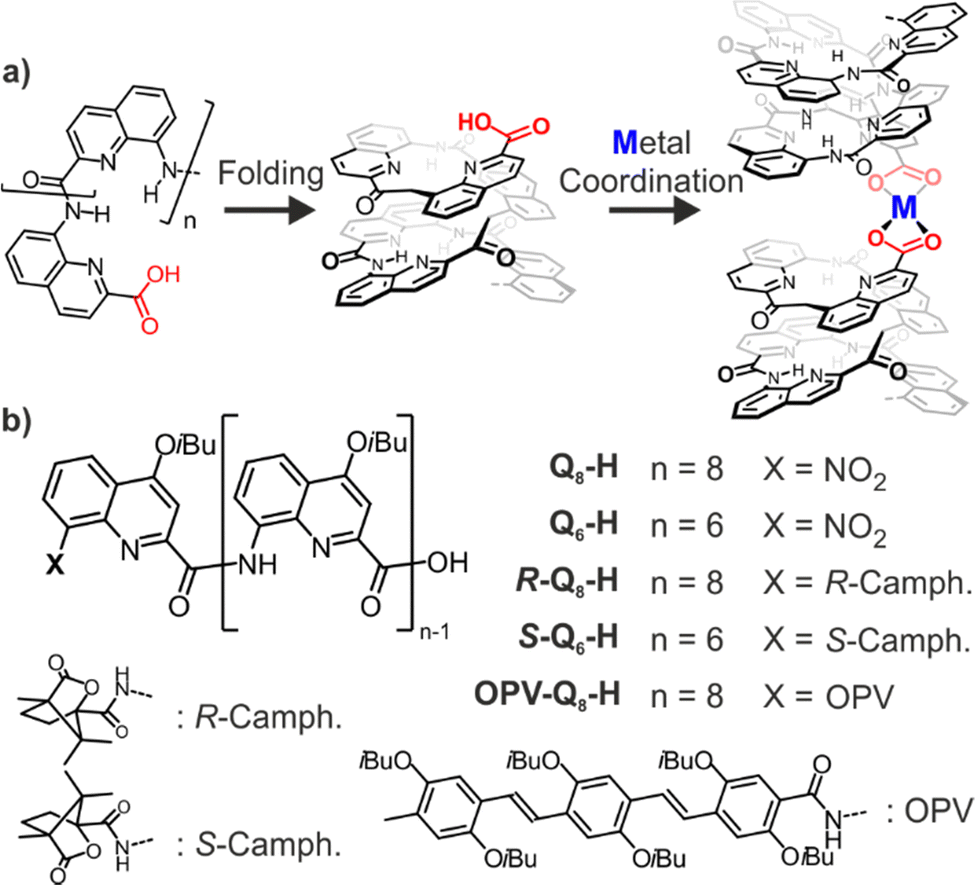 | ||
| Fig. 1 (a) Schematic representation of the folding of a helical oligoamide quinoline-based foldamer and of the formation of a dimeric complex by metal coordination. (b) Chemical formula of studied compounds. | ||
In order to better characterize this new ligation, an acid-functionalized quinoline octamer (Q8-H, Fig. 1) was synthesized based on a well-established protocol.21 Different methods were explored to prepare the desired metal complexes with this foldamer. In the presence of an excess of NaOH introduced either as a solid suspension or in an aqueous phase, the complex could be detected in a CDCl3 solution of Q8-H as evidenced by the analysis of the 1H NMR spectrum. A new set of proton signals gradually appeared at the expense of those of the initial Q8-H (Fig. S1, ESI†) which, according to diffusion-ordered spectroscopy (DOSY) NMR spectra (Fig. S7, ESI†), correspond to a bigger entity than the initial monomeric species. The new signals appeared at lower chemical shifts compared to the initial signals, indicating a strong shielding effect, in agreement with the formation of a stacked duplex. For example, characteristic NH quinoline amide signals and singlets of protons in position 3 of the quinolines were shifted downfield by around 1 ppm in the complex (Fig. 2a and b) and are in the range of those observed for the quinoline hexadecamer.25 Notably, only one set of signals is observed for the complex, indicating a completely stereoselective assembly of the helices involved. Indeed, helices from a quinoline octamer have a slow helical inversion rate25 and the NMR spectra are expected to show different signals for complexes composed of helices of similar or opposite helical handedness (PP/MM vs. PM for dimers)(Fig. S8, ESI†).
 | ||
| Fig. 2 Downfield region of 1H NMR spectrum of a CDCl3 solution (300 MHz, 298 K, 2 mM) of (a) Q8-H (b) Q8-Na-Q8 (c) Q8-K-Q8 (d) Q8-Li-Q8 (e) Q8-Ag-Q8 (f) Q8-Hg-Q8, brackets indicate the amide proton region and proton in position 3 of the quinoline units. | ||
The solid-state structure, obtained by X-ray diffraction analysis of single crystals, confirmed the formation of a Q8-Na-Q8 dimer (Fig. 3a and d). The two helically folded quinoline octamers have the same handedness and are connected through their carboxylate end group to one sodium cation in a head-to-head fashion with a perfect coaxial arrangement of the helices. The cation is hexa-coordinated to one carboxylate oxygen atom and the endocyclic nitrogen atoms of the two adjacent quinoline rings of each oligomer. The O–Na bonds are 2.23 and 2.25 Å-long, while the N–Na distances range from 2.69 to 3.05 Å (Fig. S29 and Table S2, ESI†). To fulfill the charge balance, a second sodium cation is located close to the end of the helices but outside the coaxial dimer. It is presumably not involved in the complex stabilization in solution.
 | ||
| Fig. 3 X-ray structures of dimer complexes. Side view of (a) Q8-Na-Q8 (b) Q8-K-Q8 (c) Q8-Hg-Q8. Top view of (d) Q8-Na-Q8 (e) Q8-K-Q8 (f) Q8-Hg-Q8. Foldamers are shown in sticks, whereas metal cations are displayed as balls. H atoms, side chains, solvent molecules and ions have been removed for clarity. Dashed lines are indicative of the middle of the dimeric structure and do not represent crystallographic symmetry elements. | ||
The dimerization of Q8H with NaOH was further studied by time-resolved fluorescence anisotropy (TRFA), which provided a reliable way to evaluate oligomer size and shape. To this end, Q8H was modified at its N-terminus with a trimeric oligo(phenylene vinylene) to yield the fluorescently labelled OPV-Q8H (Fig. 1b).26 Solutions of OPV-Q8H were prepared in chloroform at concentrations ranging from 13 nM to 830 μM, thus spanning five orders of magnitude. TRFA yielded the average rotational time 〈ϕ〉 of the objects generated in the OPV-Q8H solutions after adding 0.1 g of 16 M NaOH solution. 〈ϕ〉 was plotted as a function of OPV-Q8H concentration in Fig. S32 (ESI†). Before exposure to NaOH, 〈ϕ〉 was constant and measured at 0.79 (± 0.02) ns for the entire concentration range. This value is similar to the 0.78 ns determined earlier for the methyl ester of OPV-Q8H.26 The addition of NaOH led to a sigmoidal increase in 〈ϕ〉 with increasing OPV-Q8H concentration, going from 0.77 (±0.01) ns at [OPV-Q8H] < 46 μM to a constant value of 1.40 (±0.02) for [OPV-Q8H] > 100 μM. Since 〈ϕ〉 is directly related to the length of the molecular edifice, its increase indicated the formation of a larger object in solution. Based on an earlier calibration for these methyl ester quinoline oligomers,26 a 1.40 ns rotational time would be equivalent to a foldamer made of 19 quinolines, slightly larger than the 16 quinolines of an OPV-Q8-Na-Q8-OPV dimer. The difference in the expected and actual quinoline numbers (19 vs. 16) is presumably due to the second OPV moiety in the dimers since the calibration curve was established with oligomers bearing a single OPV label. Furthermore, the sigmoidal 〈ϕ〉-vs.-[OPV-Q8H] profile in Fig. S33 (ESI†) is a clear indication of a finite (discrete) association mechanism matching the dimerization of OPV-Q8H rather than non specific association.
The finding that a strong base like NaOH promoted dimerization led to study the effect of less basic salts on dimerization. The Q8-Na-Q8 dimer complex was quantitatively produced by treatment of a Q8-H solution in chloroform with sodium carbonate, although with slower kinetics (Fig. S4, ESI†). The Na+ dimeric complex was also generated with sodium acetate but not quantitatively, even in presence of a significant excess of the base for a week (Fig. S6, ESI†). The same 1H NMR spectrum was obtained for the complexes regardless of the counteranion used, suggesting that it is absent from (or had little effect on) the dimer structure in the final solution.
Having optimized the preparation of the Na+ dimer complexes, we then explored whether other metal salts could also produce dimeric complexes. Q8-H was mixed with various basic salts, and complexation was monitored by 1H NMR. Dimeric complexes were successfully prepared with K+, Li+, Ag+, and Hg2+ salts (Fig. 2c–f). Potassium complexes were formed quantitatively when treating Q8-H with an excess of either potassium hydroxide or carbonate (Fig. 2c and Fig. S9–S11, ESI†). Single crystals of the K+ dimer suitable for X-ray diffraction analysis were obtained by diffusion of n-hexane to a chloroform solution of the dimer through a dichloromethane buffer solution. The structure of the complex was found to be almost superimposable to that of the Na+ dimer (Fig. 3 and Fig. S33, ESI†). One K+ atom is connecting two helices of the same handedness, and another metal ion sits outside the coaxial dimer and fulfils the charge balance of the complex (Fig. S31, ESI†). Q8-H also afforded the Q8-Li-Q8 complex with LiOH, whereas forming Ag+ or Hg2+ dimeric complexes proceeded with acetate salts (Fig. S12–S18, ESI†). 1H NMR spectra of each metal complex presented subtle but significant differences (Fig. 2) depending on the nature of the metal used, indicating that the metal has an influence on the chemical environment of the protons of the dimeric structures. Unlike other metals, the 1H NMR spectrum of the Hg2+ complex showed twice as many signals as the other complexes indicating a loss of symmetry (Fig. 2f). This puzzling phenomenon could be explained by close inspection of the solid-state data of the complex (Fig. 3c). While the X-ray structure indicates that the helices backbones of Q8-Hg-Q8 are arranged in the same manner as for the Na+ and K+ complexes, the Hg2+ cation is not positioned at the same position as with alkaline metals. Instead it is closer to one of the two helices rendering the dimer complex dissymmetric. Furthermore, the 1H NMR spectrum of Q8-Hg-Q8 shows a set of minor amide proton signals above 11 ppm (Fig. 2f and Fig. S16 and S17, ESI†). These signals do not match those of Q8-H and likely belong to a Q8-Hg+ monomeric species.
Competition experiments were performed by mixing a solution of Q8-H with equimolar amounts of two metal salts. They revealed the following relative stabilities of the Q8-M-Q8 complexes and highlighted the prevalence of Q8-Ag-Q8: KAg > KNa ≈ KK > KLi (Fig. S19–S21, ESI†). The mercury salt was not tested because of its more complicated signal pattern.
The dynamics and possible ligand exchange in the dimeric complexes were investigated by adding a shorter quinoline hexamer, Q6-H, to a preformed Q8-Na-Q8 dimer solution in the presence of excess NaOH. The 1H NMR spectrum of the mixture revealed the formation of the expected symmetric Q6-Na-Q6 homodimer and the dissymmetric Q8-Na-Q6 heterodimer (Fig. S23–S25, ESI†). The rapid formation of the heterodimer indicates that the metal–ligand bond in the initial Q8-Na-Q8 dimer is kinetically labile. This insight led to the investigation of the stereoselectivity of the assembly observed in the crystal and presumed to also exist in solution (i.e only helices of similar handedness can form dimers). For this purpose, quinoline oligomers of 8 and 6 units bearing a chiral camphanic group of different chirality at the N-terminus were prepared to yield R-Q8-H and S-Q6-H. This chiral group is known to bias quantitatively the helical handedness of quinoline foldamers in favour of P and M helices for R- and S-camphanic groups, respectively.27 Similarly to the non-chiral analogues, 1H NMR spectra of these individual oligomers in the presence of NaOH indicated the formation of one dimer as a new set of peaks was observed in the 1H NMR spectra. However, when a mixture of R-Q8-H and S-Q6-H was exposed to NaOH, only S-Q6-Na-S-Q6 and R-Q8-Na-R-Q8 homodimers were observed and no S-Q6-Na-R-Q8 heterodimer was detected. The heterodimer formation is prevented by the opposite handedness of R-Q8 and S-Q6. This result demonstrates the chiral narcissistic self-sorting ability of this assembly for which only two helices of identical handedness can coordinate the same metal ion (Fig. S27 and S28, ESI†).
The strong carboxylate-induced complexation led to the proposal that a symmetric foldamer functionalized with carboxylic acid groups at both ends could form either dynamic polymers or macrocyclic structures upon treatment with NaOH or AcOAg. Such a symmetric foldamer, HQ2PQ2H (Fig. 4a), containing a central pyridine dicarbonyl unit flanked by two quinolines on each side was prepared. The introduction of hexyl and decyl side chains on the quinoline and pyridine units was crucial for solubility in non polar solvents. After treatment of a chloroform solution of HQ2PQ2H with NaOH, the 1H NMR spectrum showed extensive line broadening attributed to the formation of a statistical mixture of oligomers or polymers of variable length (Fig. S35, ESI†). To exert better control over the degree of polymerization, stoppers (i.e. monoacids) were introduced to cap the polymer chains and impose a stoichiometric control over the average polymer length. The titration of Q8-Na-Q8 with increasing amounts of HQ2PQ2H in the presence of NaOH was followed by 1H NMR. After the addition of 1 eq. of the diacid, new sets of peaks were observed (Fig. S36, ESI†). These were attributed to the insertion of one or more diacids to form new oligomeric complexes composed of Q8 oligomers at each end and one or more HQ2PQ2H units in the core, each connected by Na+ ions. As expected, the amide proton signals of the longer species were shifted upfield. The formation of a statistical mixture of oligomers of various lengths is reflected by the complexity and the number of news signals observed even after the addition of 0.5 equivalents. With the further addition of HQ2PQ2H, the signals of the initial Q8-Na-Q8 dimer decreased, indicating that Q8 acted as a stopper in the formation of longer structures.
 | ||
| Fig. 4 (b) CD spectra of the titration of a dry CHCl3 solution of R-Q8H (1 mM, path length 0.1 mm) with increasing amounts of HQ2PQ2H in the presence of AcOAg in excess. (c) Plot of the CD intensity at 390 nm depending on the number of equivalents of Q2PQ2 added. | ||
Circular dichroism (CD) was then applied to probe the elongation of the polymer chains upon insertion of the diacids. The chiral R-Q8-H octamer was used as stopper to dictate the selective assembly of helices of similar handedness. The P/M equilibrium of the free HQ2PQ2H is fast and was expected to shift in favor of P-helicity upon insertion in coordinated polymer chains capped with R-Q8. The P-helix selective incorporation of HQ2PQ2H in coordinated helical chains was evidenced by the amplification of the CD signal as increasing amounts of the racemic diacid was added. Thus, HQ2PQ2H was added to a 1 mM solution of R-Q8-H in the presence of an excess of silver acetate, the best mediator for self-assembly. Such a high concentration of stoppers was used to ensure a strong complexation. Increasing amounts of non-chiral diacid produced a continuous increase of the CD signal after equilibration (Fig. 4b and c). Due to the high initial concentration, the CD signal was saturated after the addition of ca. 17 equivalents of diacid. The length of the polymer could be estimated from the intensity of the CD signal, which is proportional to the number of quinolines in the oligomer. Assuming that the initial CD signal was due to the 16 quinolines of the initial dimer, the 2.2-fold increase in CD signal indicated that addition of 8 equivalents of diacid generated polymers with 35 quinolines on average, corresponding to the insertion of 4.8 diacids. This metallopolymer represents a foldamer of considerable length that would be difficult to obtain by traditional synthetic methods.
In conclusion, this work describes a novel coaxial ligation of helical foldamers upon metal coordination. Two helical strands possessing one acid termini each were bound by a cation in a head-to-head fashion. The metal coordination transferred the handedness of one foldamer to the other. The use of a bis-acid functionalized foldamer resulted in the formation of supramolecular metallopolymers that were characterized by CD and NMR. The stability of these complexes in organic solvents, the variety of metals that can be used in the assembly, the dynamics of the ligand exchange and the stereoselectivity of the process make this carboxylate-induced ligation of aromatic foldamers a powerful method to create long rods that can find uses in the context of water channeling12,13 or charge transport properties.7 Further studies on the rigidity of these supramolecular polymers and their thermodynamic stability are in progress and will be reported in due course.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173 CrossRef CAS.
- S. Hecht and I. Huc, Foldamers: Structure, Properties and Applications, 2007 Search PubMed.
- V. Corvaglia, I. A. M. Amar, V. Garambois, S. Letast, A. Garcin, C. Gongora, M. D. Rio, C. D-Sabourin, N. Joubert, I. Huc and P. Pourquier, Pharmaceuticals, 2021, 14, 624 CrossRef CAS PubMed.
- C. Tsiamantas, S. Kwon, C. Douat, I. Huc and H. Suga, Chem. Commun., 2019, 55, 7366 RSC.
- M. Vallade, M. Jewginski, L. Fischer, J. Buratto, K. Bathany, J.-M. Schmitter, M. Stupfel, F. Godde, C. D. Mackereth and I. Huc, Bioconjugate Chem., 2019, 30, 54 CrossRef CAS PubMed.
- X. Li, N. Markandeya, G. Jonusauskas, N. D. McClenaghan, V. Maurizot, S. A. Denisov and I. Huc, J. Am. Chem. Soc., 2016, 138, 13568 CrossRef CAS PubMed.
- A. Mendez-Ardoy, N. Markendeya, X. Li, Y.-T. Tsai, G. Pecastaings, T. Buffeteau, V. Maurizot, L. Muccioli, F. Castet, I. Huc and D. M. Bassani, Chem. Sci., 2017, 8, 7251 RSC.
- S. I. Stupp and L. C. Palmern, Chem. Mater., 2014, 26(1), 507 CrossRef CAS.
- C. Tsiamantas, X. de Hatten, C. Douat, B. Kauffmann, V. Maurizot, H. Ihara, M. Takafuji, N. Metzler-Nolte and I. Huc, Angew. Chem., Int. Ed., 2016, 55, 6848 CrossRef CAS PubMed.
- S. De, B. Chi, T. Granier, T. Qi, V. Maurizot and I. Huc, Nat. Chem., 2018, 10, 51 CrossRef CAS PubMed.
- X. Yan, P. Weng, D. Shi and Y.-B. Jiang, Chem. Commun., 2021, 57, 12562 RSC.
- Y. Huo and H. Zeng, Acc. Chem. Res., 2016, 49, 922 CrossRef CAS PubMed.
- J. Shen, R. Ye, A. Romanies, A. Roy, F. Chen, C. L. Ren, Z. Liu and H. Zeng, J. Am. Chem. Soc., 2020, 142, 10050 CrossRef CAS PubMed.
- Y. Dai, T. J. Katz and D. A. Nichols, Angew. Chem., Int. Ed. Engl., 1996, 35, 2109 CrossRef CAS.
- J. L. Greenfield, F. J. Rizzuto, I. Goldberga and J. R. Nitschke, Angew. Chem., Int. Ed., 2017, 56, 7541 CrossRef CAS PubMed.
- C. Nuckolls, T. J. Katz, G. Katz, P. J. Collings and L. Castellanos, J. Am. Chem. Soc., 1999, 121, 79 CrossRef CAS.
- H. Ito, M. Ikeda, T. Hasegawa, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2011, 133(10), 3419–3432 CrossRef CAS PubMed.
- D. Bindl, P. K. Mandal, L. Allmendinger and I. Huc, Angew. Chem., Int. Ed., 2022, 61, e202116509 CrossRef CAS PubMed.
- L. A. Cuccia, E. Ruiz, J.-M. Lehn, J.-C. Homo and M. Schmutz, Chem. – Eur. J., 2002, 8, 3448 CrossRef CAS PubMed.
- W. Cai, G.-T. Wang, P. Du, R.-X. Wang, X.-K. Jiang and Z.-T. Li, J. Am. Chem. Soc., 2008, 130, 13450 CrossRef CAS PubMed.
- T. Qi, T. Deschrijver and I. Huc, Nat. Protoc., 2013, 8, 693 CrossRef PubMed.
- N. Ahmad, A. H. Chughtai, H. A. Younus and F. Verpoort, Coord. Chem. Rev., 2014, 280, 1 CrossRef CAS.
- G. E. Decker, G. R. Lorzing, M. M. Deegan and E. D. Bloch, J. Mater. Chem. A, 2020, 8, 4217 RSC.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O’Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS PubMed.
- N. Delsuc, T. Kawanami, J. Lefeuvre, A. Shundo, H. Ihara, M. Takafuji and I. Huc, Chem. Phys. Chem., 2008, 9, 1882 CrossRef CAS PubMed.
- J. Wang, H. Little, J. Duhamel, X. Li, N. Markandeya, V. Maurizot and I. Huc, Macromolecules, 2019, 52(15), 5829 CrossRef CAS.
- A. M. Kendhale, L. Poniman, Z. Dong, K. Laxmi-Reddy, B. Kauffmann, Y. Ferrand and I. Huc, J. Org. Chem., 2011, 76, 195 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General synthetic experimental details, NMR spectra, fluorescence anysotropy and X-ray analysis. CCDC 2194086 (Q8-Na-Q8), 2194087 (Q8-K-Q8), 2194085 (Q8-Hg-Q8). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc00866e |
| This journal is © The Royal Society of Chemistry 2023 |