
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBeyond DNA: new probes for PAINT super-resolution microscopy
Marrit M. E.
Tholen
 a,
Roderick P.
Tas
bc,
Yuyang
Wang
cd and
Lorenzo
Albertazzi
*a
a,
Roderick P.
Tas
bc,
Yuyang
Wang
cd and
Lorenzo
Albertazzi
*a
aDepartment of Biomedical Engineering, Institute of Complex Molecular Systems, Eindhoven University of Technology, Eindhoven, The Netherlands. E-mail: l.albertazzi@tue.nl
bDepartment of Chemical Engineering and Chemistry, Laboratory of Self-Organizing Soft Matter, Eindhoven University of Technology, Eindhoven, 5612 AP, The Netherlands
cInstitute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
dDepartment of Applied Physics, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
First published on 29th May 2023
Abstract
In the last decade, point accumulation for imaging in nanoscale topography (PAINT) has emerged as a versatile tool for single-molecule localization microscopy (SMLM). Currently, DNA-PAINT is the most widely used, in which a transient stochastically binding DNA docking–imaging pair is used to reconstruct specific characteristics of biological or synthetic materials on a single-molecule level. Slowly, the need for PAINT probes that are not dependent on DNA has emerged. These probes can be based on (i) endogenous interactions, (ii) engineered binders, (iii) fusion proteins, or (iv) synthetic molecules and provide complementary applications for SMLM. Therefore, researchers have been expanding the PAINT toolbox with new probes. In this review, we provide an overview of the currently existing probes that go beyond DNA and their applications and challenges.
 Marrit M. E. Tholen | Marrit M. E. Tholen obtained her BSc in Medical Sciences and Technology (2017) and her MSc in Biomedical Engineering (2019) at Eindhoven University of Technology. As part of her master's degree, she worked on the development of polymersomes for mucosal vaccines and performed an internship at the Radboud University Medical Centre (Nijmegen, The Netherlands) where she worked with gelatin colloidal gels. In 2020, she joined the Nanoscopy for Nanomedicine group for her PhD, where she is combining research with teaching. She is working on the development of probes for characterization of nanoparticles using super-resolution microscopy techniques. |
 Roderick P. Tas | Roderick Tas obtained his MSc degree in Molecular and Cellular Life Sciences (2014) and a PhD in Biophysics (2019) from Utrecht University (UU, The Netherlands). He then joined TU/e as a postdoctoral researcher. He was awarded a NWO-VENI fellowship in 2020 and became fellow of the Institute for Complex Molecular Systems (ICMS) in 2021. In 2023 he was appointed Assistant Professor in the department of Biomechanical Engineering at Delft University of Technology. During his studies Roderick has combined molecular biology, biochemistry and biophysics to achieve molecular understanding and visualisation of complex biological processes. Now he continues this track to understand the interactions between cells and engineered materials. |
 Yuyang Wang | Dr Yuyang Wang is the microscopy facility manager at the ICMS (TU/e). He obtained his PhD in biophysics for his work on single-molecule fluorescence and nanoplasmonics at TU/e. From 2020 he continued his research in biophysics at TU/e after being awarded a joint postdoctoral fellowship from Institute for Complex Molecular Systems (ICMS, TU/e) and Institute for Bioengineering of Catalonia (IBEC). His current research activities are optical super-resolution microscopy (SRM), correlative light-electron microscopy (CLEM) and nanoplasmonics-based optical sensing. |
 Lorenzo Albertazzi | Dr Lorenzo Albertazzi obtained his PhD in Biophysics (2011) from Scuola Normale Superiore (Pisa, Italy). He then joined TU/e as a postdoctoral researcher. In 2013, he was awarded a fellowship at the ICMS, and in 2014 he became a NWO/VENI fellow. In 2015, he moved to IBEC to start the ‘Nanoscopy for Nanomedicine’ group. In 2018, he was appointed Associate Professor at the TU/e department of Biomedical Engineering. By combining chemistry and biophysics, he is aiming to achieve a molecular understanding of synthetic materials in the biological environment, using optical microscopy and nanoscopy. |
Introduction
Super-resolution microscopy enabled researchers to study biological processes and synthetic materials with diffraction-unlimited precision. Specifically, single-molecule localization microscopy (SMLM) has been used extensively to obtain a ten-fold increase in resolution by exciting individual fluorophores sequentially, detecting them and reconstructing their position (and dynamics) with nanometric precision. For biological samples, SMLM opened the possibility for imaging cellular ultrastructures below the diffraction limit1–3 leading to key discoveries such as the periodic cytoskeletal structure in axons.1 SMLM is not only useful in biological sciences, but also in materials sciences. For example, the exchange of monomers in supramolecular fibres was quantified, which unravelled mechanisms on the single-molecule scale that were not experimentally proven before.4SMLM can be further divided into three main families depending on the mechanism of single fluorescent molecule separation: (i) photoactivation/switching based microscopy, including photo-activated localization microscopy (PALM), stochastic optical reconstruction microscopy (STORM), direct STORM (dSTORM); (ii) dynamic labelling microscopy including point accumulation for imaging in nanoscale topography (PAINT) based on reversible binding, and (iii) fluorescence life-time separation based microscopy. All these methods, based on different separation methods, have their own advantages and are useful in both biology and materials science, as thoroughly reviewed elsewhere.5–11 Among them, PAINT is growing in interest thanks to the practicality of dynamic labelling in various samples. In this perspective, we aim to provide insights into the state of the art in probe design for PAINT microscopy and extending the toolbox beyond DNA as probes.
The concept of PAINT is based on the premise that fluorescent probes, targeted to a molecule of interest freely diffusing through the solution (Fig. 1, middle panel). Upon binding to the target, they are immobilized, and the fluorescent signal of the single-molecule appears on the camera, which can be localized through a fitting procedure. As the kinetics of the probe ensure unbinding, the fluorescent signal is turned off again until a new molecule binds. As the probes are replenished continuously, it is not sensitive to photobleaching, which is a major advantage of this method over other SMLM techniques. This allows for long imaging times and thus a higher accuracy. Moreover, by combining multiple probes with different dyes, multiplexing can be achieved.12 In addition to high resolution and reconstruction in all three dimensions, PAINT allows for the quantitative analysis of synthetic and biological materials whilst maintaining the sample integrity.8,13 In recent years, the technique has evolved to become a standard technique, and its potential has interested a broad range of scientists across various disciplines as was represented by numerous reports and reviews.5,6
 | ||
| Fig. 1 An overview of the probes discussed in this paper. In the middle, the general idea behind PAINT approaches is illustrated. The four main categories for probes are: endogenous probes, engineered probes, fusion-based probes and synthetic probes. Schematics created with Biorender.com. | ||
The first study based on this PAINT principle used the hydrophobic dye Nile Red that transiently binds hydrophobic regions.14 Using this probe, Sharonov et al. (2006) showed the potential of transiently binding fluorescent probes into large unilamellar vesicles (LUVs) and supported bilayers, to separate the fluorophores in space and time, resulting in a super-resolution image. More recently, a key step towards the widespread adoption of PAINT involved the development of DNA-PAINT in 2010.12 DNA-PAINT relies on two complementary single-strand DNA sequences that can transiently bind to each other: a “docking” strand bound to the target of interest and an “imaging” strand that act as an affinity probe.12 One of the major advantages of this DNA-based PAINT approach is that the interaction is highly tuneable in specificity and affinity by modifying the base–pair sequence and length. As a result, acquisition times can be fine-tuned, and multicolour imaging can be achieved by having a mixture of specific docking/imaging pairs. While Nile Red only allows for imaging hydrophobic materials, DNA can be used to tag a wide variety of molecules using intermediate targeting agents (i.e. antibodies, nanobodies, and affimers), opening up the possibility of imaging a lot more characteristics of biological and synthetic objects. Nowadays, multiplexing can be achieved by sequential imaging up to 124 colours, by rational design of docking and imaging strands.15 Moreover, by concatenating sequences, the speed can be increased by 100-fold.16 From the data, the specific number of binding events can be derived and by using the mean dark time (τoff) and association rate (kon) of the DNA pair, the binding events can be translated to number of molecules.17 This method, called qPAINT, is an established complementary method to DNA-PAINT. Therefore, DNA-PAINT is the current standard in PAINT microscopy.
The high flexibility of DNA is the strong point of this method, but using DNA also comes with pitfalls. Imaging strands give rise to non-specific binding events due to charge interactions, which complicates data interpretation.18 Moreover, the low stability of the DNA inside cells and labelling of targets inside living cells provide obstacles for live-cell imaging.19 For this last hurdle, labelling of the target of interest with a docking strand is required, which usually means that an endogenous molecule or antibody is modified with a small DNA strand, which perturbs the cell or tissue. This introduces an intermediate affinity probe, meaning that the measurements are indirect. Although DNA-PAINT is still the golden standard, there is a need to expand the current PAINT toolbox with new probes that tackle these challenges and complement the DNA-based probes.
In this review, we will shine light on the current status of probes for PAINT that go beyond DNA, and how these probes have a high potential for new PAINT applications. First, we will discuss what should be considered when one wants to develop a new probe for PAINT applications. Thereafter, we will divide the existing probes into four main categories: (i) endogenous targeting molecules, such as sugars, (ii) engineered probes, such as aptamers, (iii) fusion protein-based probes and (iv) synthetic probes (see Fig. 1). Furthermore, we will provide a perspective on the future of PAINT. We hope that this review can act as inspiration for researchers in pursuit of the development and use of DNA-free PAINT probes.
Challenges for probe design
The beauty of DNA-PAINT is its programmability. As discussed, by adjusting the DNA sequence of the designed probe, one can change the affinity between the pair and therewith the acquisition speed. When one wants to design a new PAINT probe for super-resolution microscopy, without the use of DNA, a few considerations have to be taken into account.The first consideration is that the probe-target affinity (defined by kon and koff) must be in the right range. To achieve effective PAINT, the association and dissociation should take place at a rate that is within a certain range, with a rule of thumb that the equilibrium dissociation constant (eqn (1)) lies between 100 nM and 1 μM.
 | (1) |
Weaker interactions (high Kd) may lead to too little or too short events, for SMLM, whereas stronger interactions (low Kd) make it harder to observe single interactions due to long binding times (as illustrated in Fig. 2(A)). For weaker interactions, it might be possible to increase the concentration in order to increase the chances of binding. Moreover, once the probe binds, the interaction will be very short (high koff) and will not be detected or detected inaccurately. In the case of too strong interactions, the point spread functions of the binding events will overlap and single-molecule will be hard to distinguish. Moreover, saturation of the target molecules may lead to error in counting. Potentially, one could lower the concentration of probe to overcome this issue but this would result in a significant extension of measurement time. The affinity requirement must function as a guideline to design probes for a direct PAINT approach. Endogenous biological interactions span many orders of magnitude in terms of affinity, with Kd ranging from fM (e.g. streptavidin-biotin) to high μM (e.g. some sugar–lectin interactions) and only a subcollection of them are usable for PAINT. In the case of engineered probes, it is important to target the right affinity range without sacrificing too much specificity. These challenges must be tackled to go beyond DNA-based PAINT probes.
 | ||
| Fig. 2 Schematic overview of the considerations that have to be made while designing or choosing a PAINT probe. (A) The affinity of the probe towards its target. Too weak will give no signal or the acquisition time is long and too strong will not result in single molecule binding. (B) The choice of fluorophores is important. The color and photophysical properties are of importance as is the buffer conditions in which is being imaged. (C) For imaging in cells, a choice between live and fixed cells has to be made. Furthermore, the permeability and the stability of the probe should be optimized. During imaging, the pool of probes should be replenished and the lasers should not be toxic to live cells. Schematics created with Biorender.com. | ||
The second consideration is the choice of fluorophores, based on their photophysical properties, summarized in Fig. 2(B). In SMLM, the localization precision is limited by the Cramér–Rao lower bound (CRLB, eqn (2)), where σ0 is the standard deviation of the point-spread function (PSF), and N the photon budget collected by the camera.
 | (2) |
A higher number of photons detected from one event increases the localization precision, while photostability poses a threat for long-time measurement with a high precision. To overcome this, specific imaging buffer compositions have been used, to prevent bleaching and enhance photostability by using an oxygen-scavenging system and a triplet-state quencher. As a native advantage, the PAINT signal can be replenished after bleaching by a large pool of probes in solution.20,21 ATTO and Janelia fluor dyes, known for their photostability, are suitable dyes that overcome most of these aforementioned issues.22,23 It might be worth using fluorescent proteins (FPs) as the stoichiometry is controlled, the labeling efficiency is 100% and they are less prone to aspecific binding than DNA.24–26 FPs have typically worse photophysical properties than organic dyes but new generation of mutants have massively improved brightness and stability and are promising candidates for PAINT.
The third consideration is in cellular systems including live and fixed cells, where PAINT imaging is performed. We outline the cellular challenges in Fig. 2(C): (1) membrane-permeability of the probes, (2) intracellular probe pool, (3) probe stability in cells, and (4) probe toxicity and intrusiveness in cells. These challenges present themselves differently between live and fixed cells. In fixed cells membrane permeabilization for probes is possible, allowing them to reach intracellular targets as well as to equilibrate the intracellular pool of probes with the external one. Though not sensitive to probe toxicity, cell fixation alters epitope presentation and cell ultrastructure and could impact the affinity of probes designed to target native proteins. In live cells, probe design is challenging, because only membrane-permeable probes can be used, limiting the choice of fluorophores to small and hydrophilic molecules. Genetically encoded tags have been used for PAINT but inside the cell, the intracellular pool is not infinite anymore and the acquisition time can be limited.27 Moreover, lowering the laser power might be needed to prevent both bleaching of the probes and phototoxicity, but results in a lower photon budget and therefore lower resolution. Moreover, live cell metabolism may degrade the probes, which is a large hurdle in the case of DNA in DNA-PAINT because of the presence of DNAses. Degradation of these probes has limited the targets to extracellular proteins at the membrane in live-cells.19 Combined, there are a wide range of parameters to consider while designing a new probe for PAINT. Nevertheless, there is still a plethora of options to choose from. In the next sections the state-of-the-art and future perspectives of the four families of DNA-independent probes for PAINT are discussed.
Endogenous targeting molecules
One approach for the development of highly specific PAINT probes without the need for DNA is using endogenous purified, fluorescently labelled, transient interaction partners to probe a protein of interest (Fig. 3). By using such probes, the natural interactions between proteins in biology are maintained, which could give more insights into the kinetics of these interactions, but also into processes in nature. One major advantage of this approach over DNA-PAINT is that endogenous proteins can be imaged without labeling or modification of the cells, rapidly after fixation. Furthermore, no intermediate targeting probes (i.e. antibodies, nanobodies, genetic tags) are required resulting in a minimal linkage error because only the small protein fragments separate the localized fluorophore from the target. Additionally, because well-characterized interactions are used, the labelling stoichiometry is known, making this method applicable for quantitative PAINT imaging. As discussed before, one essential criterium for these probes to be suitable for PAINT acquisitions is that they need high nM to low μM affinity to the target molecule. One of the first studies that adopted this principle to perform PAINT in order to resolve cellular proteins was termed image reconstruction by integrating exchangeable single-molecule localization (IRIS).28 In this work, schematically represented in Fig. 3(A), the authors screened well-characterized endogenous binders to various cellular targets in fixed cells. By testing purified fragments of well-known protein interactors to actin, microtubules, focal adhesions and intermediate filaments, they were able to find transient binders to resolve these structures with nanometric accuracy using the PAINT principle and demonstrate the potential of proteins as alternatives to DNA.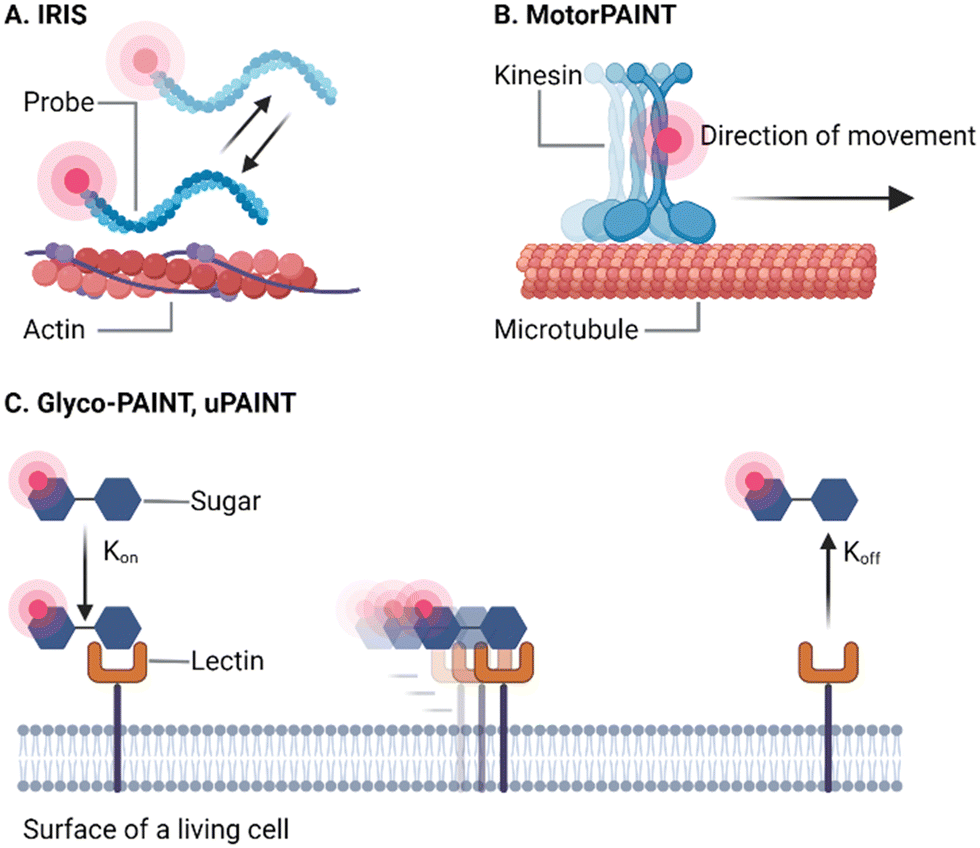 | ||
| Fig. 3 Three examples of endogenous probes as discussed above. (A) IRIS uses (fractions of) endogenous transient interaction partners to the target of interest, e.g. the lifeAct fragment coupled to a fluorophore for PAINT acquisition of actin filaments. (B) Motor-PAINT uses purified active kinesins to reconstruct the microtubule cytoskeleton and directly infer the orientation of the microtubules. (B) Glyco-PAINT and uPAINT are live-cell imaging methods that can be used to track the motion of receptors on the cell membrane. The probe binds to the receptor with a certain kon, moves along with the receptor, until it is released again (koff). Schematics created with Biorender.com. | ||
Quantitative PAINT imaging was demonstrated by another study using the PIPKY fragment, characterizing the number of talin in focal adhesions.29 Similar to IRIS, another study used well-known downstream signaling proteins to perform protein-PAINT of several membrane-bound signaling proteins such as the T-cell receptor. Purification of downstream signaling molecules such as the ZAP70-tSH domain, PI3K, Grb2 and other proteins, allowed the diffraction unlimited detection of signaling proteins and their specific modifications in fixed immunological synapses of T-cells that corresponded to the different stages of synapse maturation.24
Motor-PAINT (Fig. 3(B)) extends the principle of PAINT based on endogenous protein–protein interactions aimed to resolve the actin and microtubule cytoskeleton.30 By using purified motor-proteins that actively walk along extracted and fixed cytoskeletons, this approach can provide a super-resolved map of actin or microtubule cytoskeletons while simultaneously inferring the absolute polarity of the filaments in the network obtained by single-molecule tracking. As a result, the “directions” of the cellular highways can be studied for more in-depth understanding of the cellular traffic rules, because the polarity of the filaments directly dictates cellular transport by the various motor-proteins.
All examples above show that PAINT-based on endogenous interactions provides a powerful tool to resolve intracellular structures based on a one-step labelling approach. However, it should be noted that labelling using the IRIS or motor-PAINT approaches requires prior knowledge of endogenous binders and because intracellular proteins are targeted, fixation and permeabilization is required to get the protein-based PAINT probes to the structure of interest. As a consequence, screening for probes with suitable affinity after chemical fixation remains challenging and labor intensive, limiting the general applicability of the method.
PAINT via endogenous interactors can also be performed on live-cells to study the dynamics of proteins. One of the first examples for this strategy was universal PAINT (uPAINT), where the authors used low concentrations of fluorescently labelled epidermal growth factors (EGF) as ligand and imager for the dynamics of EGF-receptors, as is illustrated in Fig. 3(B). By tracking receptor dimers and comparing them to the whole receptor population, they were able to show at the single complex level that these had a higher tendency to become immobile, likely due to being targeted for endocytosis. However, so far most uPAINT applications have relied on high affinity probes such as strong ligand–receptor interactions or nanobodies31,32 and as a result, single molecule imaging is achieved only at very low concentrations rather than through transient binding.33
In our group, we developed an expansion of the concept of visualizing proteins with endogenous ligands and binders named glyco-PAINT (Fig. 3(C)).34 In this method, the mannose receptor, a crucial receptor in the immune response against pathogens, was visualized by fluorescently labelled sugar ligands. By using specific carbohydrate-chemistry, we were able to generate a library of glycan-containing probes with different repeats and conformations. This allowed us to systematically study how valency and structure affect the kinetics of the sugar–lectin interaction in live cells. Additionally, by tracking the probes upon binding, we were able to investigate how receptor diffusion and uptake depend on the ligand. Using this strategy, we found that valency, conformation and specificity are the important driving forces in determining the on-rates and receptor internalization.
uPAINT and glyco-PAINT are excellent examples of how probe based endogenous interactions can track the differential dynamics of populations of extracellular receptors at the single molecule level in living cells. The latter is a major advantage because it obtains extra information, not only about the receptor but also about the affinity between probe and target, compared to fixed cells. This comes at the cost of the ability to reconstitute a snapshot of cellular structures, due to the long integration times that are typically required. Therefore, these approaches highlight the importance of selecting the interaction strength, because whereas the high affinity interactions in uPAINT perform better in single particle tracking, PAINT reconstructions and kinetic information is better obtained using lower affinity interactions. Furthermore, whereas fixation compatibility of the interactions does not need to be considered, these probes are limited to extracellular targets due to the impermeability of the cell membrane for many conventional probes.
IRIS, motorPAINT, protein-PAINT, uPAINT and glyco-PAINT demonstrate the potential of using endogenous interactions to obtain diffraction unlimited images and dynamics with controlled labelling stoichiometry without intermediate targeting molecules. Additionally, in contrast to DNA-PAINT, physiologically relevant data regarding the binding kinetics (e.g. kon/koff) can be extracted because PAINT is performed through endogenous interactions. In the future, similar principles could be extended to many other cellular targets, but it should be noted that this approach is more labor intensive because the PAINT interaction is directly encoded in the selectivity of the probes, which needs to be tuned for transient binding every time a new target needs to be imaged.
Engineered probes
As a consequence of their often incompatible or suboptimal affinity towards their target, many endogenous binding partners are in general not suitable for PAINT applications, making the number of probes that can be developed in the previous category limited. Alternatively, selective targeting in DNA-PAINT is often achieved through highly specific, high affinity intermediate coupled to a docking strand like antibodies nanobodies or aptamers.32,35 Smart engineering of these existing probes coupled to organic fluorophores, in a way that they become transient and thus suitable for PAINT themselves, is an option that is explored for new PAINT probes (Fig. 4). This strategy opens up the possibility to fine-tune interactions between probe and target of interest. Furthermore, the probe can be designed in a way that it is cell-permeable and stable, enabling live-cell imaging. Often there are already probes developed that work in fixed cells, for bulk staining, so the starting point for the development of a probe in the PAINT regime is already defined. However, modifying these probes, specifically selected for very high affinity, to bind transiently may pose a challenge.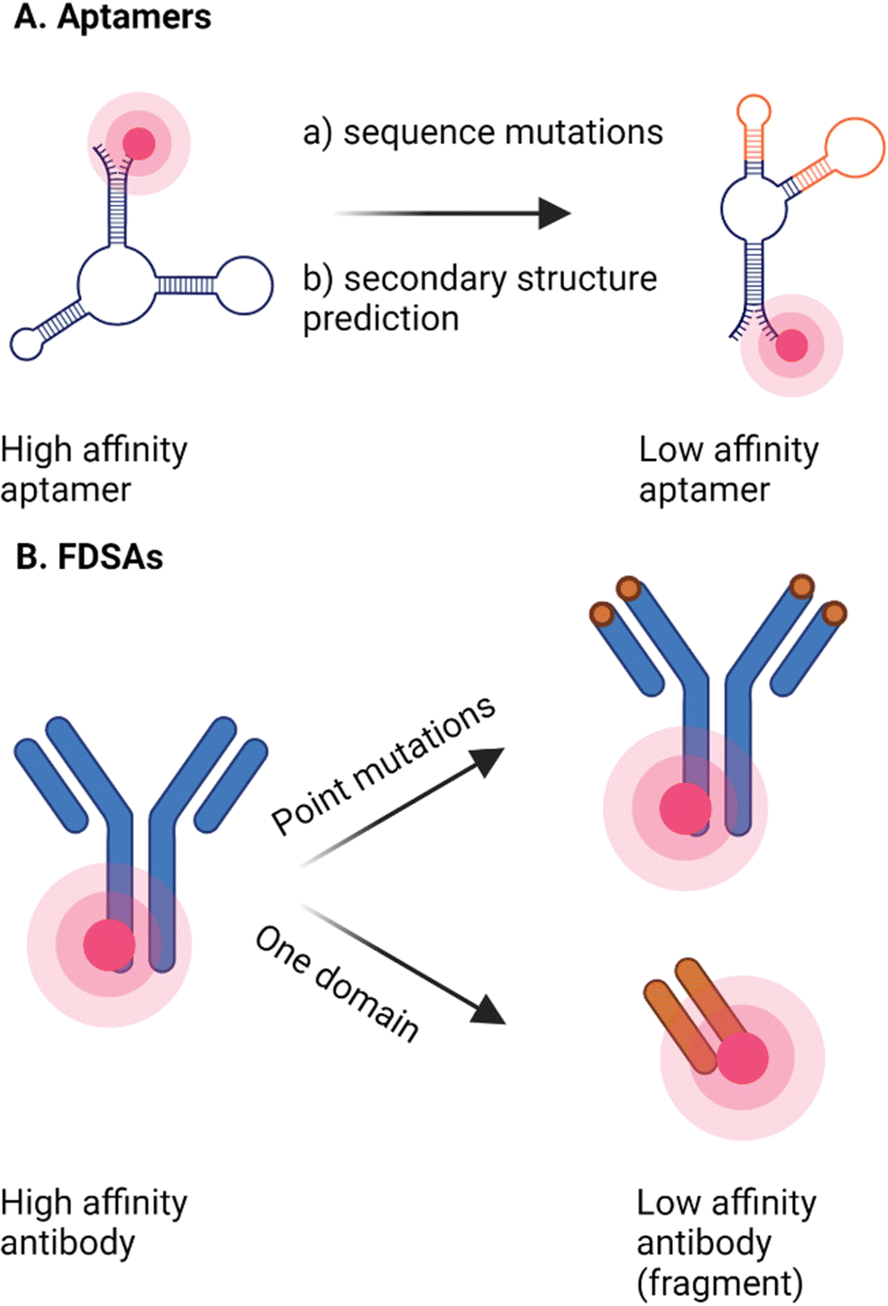 | ||
| Fig. 4 Two examples of engineered probes. (A) High affinity aptamers can be adapted by mutations in the sequence, resulting in probes with a lower affinity, but with the same specificity. (B) FDSAs are antibody fragments that are rationally engineered by introducing point mutations in the binding site. This results in antibody fragments with a lower affinity towards their target, while the specificity is maintained. Schematics created with Biorender.com. | ||
One such approach where this was achieved, is the rational engineering of aptamer sequences for transient conditions (Fig. 4(A)).36 Aptamers are short sequences of DNA or RNA artificially made to bind to a specific target molecule. While generally aptamers are selected to have a high affinity for the target, it is possible to obtain aptamers with weak and reversible binding. This can be done during the selection process or by changing the sequence to decrease the affinity of the lead aptamer selected. This way, the protein of interest can be imaged without limitations caused by photobleaching and no perturbation of the endogenous behaviour. In this case, we designed aptamers in a way that they have a tail that can bind to the complementary fluorescently labelled anti-tail, resulting in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 aptamer:fluorophore complex. As aptamers are generally not membrane-permeable, this approach is limited to membrane receptors. Not only the number and density of proteins of interest can be determined, but also the motion on the cell membrane of live cells, much like uPAINT and glyco-PAINT.36 Aptamers can also be used for RNA imaging by using fluorescent light-up aptamers (FLAPs). These aptamers are able to bind to a contact-quenched fluorophore–quencher conjugate, and upon binding the contact between the conjugate is broken and the aptamer lights up. By introducing the sequence of the aptamer in the cells by transfection, the imaging can also be performed in live cells. Furthermore, RhoBAST is a FLAP that was designed to function under physiological conditions and with excellent brightness and thermostability. Together, these characteristics make it an interesting tool for RNA imaging in both fixed and live cells, with low background and at low concentrations of target with high spatial precision.37
:1 aptamer:fluorophore complex. As aptamers are generally not membrane-permeable, this approach is limited to membrane receptors. Not only the number and density of proteins of interest can be determined, but also the motion on the cell membrane of live cells, much like uPAINT and glyco-PAINT.36 Aptamers can also be used for RNA imaging by using fluorescent light-up aptamers (FLAPs). These aptamers are able to bind to a contact-quenched fluorophore–quencher conjugate, and upon binding the contact between the conjugate is broken and the aptamer lights up. By introducing the sequence of the aptamer in the cells by transfection, the imaging can also be performed in live cells. Furthermore, RhoBAST is a FLAP that was designed to function under physiological conditions and with excellent brightness and thermostability. Together, these characteristics make it an interesting tool for RNA imaging in both fixed and live cells, with low background and at low concentrations of target with high spatial precision.37
When performing DNA-PAINT, antibodies linked to a docking strand are commonly used as the affinity probe.38,39 As antibodies themselves usually have a high affinity for their target, these proteins are not suitable for transient interactions by themselves. It was found that, when adding potassium thiocyanate (KSCN), a chaotropic agent, to a Fab domain of an anti-hemagglutinin antibody, the hydrogen bonds between target and Fab domain are disrupted, resulting in an increased dissociation rate of the Fab domain. This method was used in super-resolution census of molecular epitope tags (SR-COMET). Compared to dSTORM and DNA-PAINT, this method has a higher quantification consistency, and it does not suffer from artifacts associated with immunofluorescence staining such as steric hindrance. However, although the koff is increased by multiple orders of magnitude, the kinetics are still slow, which means that the acquisition time can still take up to a day. Additionally, the chaotropic effect is highly dependent on the amino-acid environment of the antibody, which might limit its application.40 Recently, the interest in developing fast-dissociating but at the same time highly specific antibodies for immunoassays and super resolution microscopy has increased. Miyoshi et al. (2020) have developed a pipeline in which they can screen for fast-dissociating, highly specific monoclonal antibodies (FDSAs) that can be used for image reconstruction by IRIS (Fig. 4(B)). By crosslinking Protein A/G to a glass surface, antibodies can be captured, after which affinities of these captured antibodies observed by using single molecule TIRF microscopy. The antibodies that are most suitable for the desired application can be selected. Compared to DNA-PAINT, Fab probes elevate the labelling density as they will bind to new epitopes after each dissociation round. Furthermore, during multiplexing, the new Fab probes will not be spatially interfered with by Fab probes of the previous rounds.41,42 Recently, these probes were applied to image multiple components of single cells, as a proof of principle, and on neurons. By site-directed mutagenesis, the dissociation rate of the probes was increased, without compromising in specificity. More specifically, tyrosine residues were point mutated into glycine and alanine, which resulted in an increase in koff by 100-fold, and no increase in non-specific interactions during imaging. When compared to Lifeact probes, the fast-dissociating mutants performed equally well, while the slow dissociating mutants only sketched a broad outline of the actin fibres. Combining multiple antibodies, they were able to do multiplexing of 7 different epitopes. The toolbox was expanded with two nanobodies to be able to map endogenous proteins in neurons. When compared to published dSTORM and DNA-PAINT results, IRIS antibody-probes show more continuous labelling compared to both techniques and a 4-fold higher labelling density than STORM. Unfortunately, this technique might not be applicable to all antibodies, as the development requires sequence information and cocrystal structures, which are often lacking in antibodies.43
Aptamers and engineered antibodies might pave the way to a more rational development of probes, even for endogenous proteins that have an affinity outside of the typical PAINT regime. However, screening for new suitable probes with the correct affinity is labour intensive. The aforementioned techniques were only applied to targets that have a natural high affinity for their ligand. We envision that by rationally engineering proteins with a low affinity, such as lectins, into proteins with a slightly higher affinity, the toolbox for PAINT can even be further expanded in the future.
Fusion protein-based probes
One alternative to endogenous targets or engineered intermediates, such as antibodies and nanobodies to achieve specific labelling, is PAINT imaging via fusion proteins. In this approach, proteins of interest (within the cell) are genetically fused to a “docking” peptide/protein that can interact with organic fluorophores/fluorogen imaging molecules or optimized, labelled protein interaction partners. One advantage of this approach is that, in contrast to endogenous binders, standardized interactions can be used and the labelling stoichiometry is known, making them suitable to label proteins for which no specific targets exist (Fig. 5). However, similar to the endogenous probes, the transient interactions for this approach need to be optimized to be compatible with sample preparation methods such as various fixatives. Additionally, it should be considered that recombinant fusion of the docking peptide/protein does not perturb the function of the protein of interest.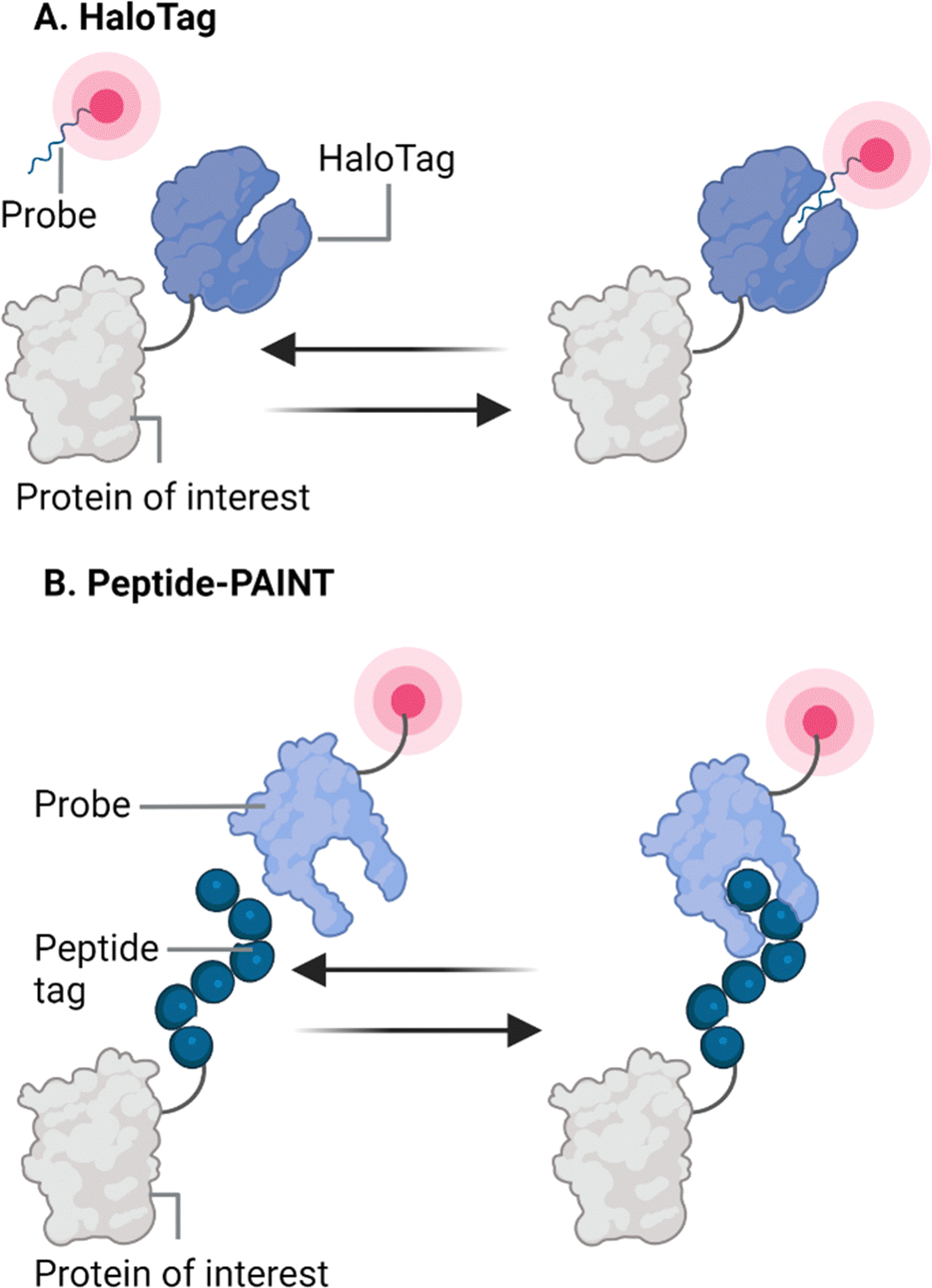 | ||
| Fig. 5 Schematic examples of two fusion-protein approaches. (A) A combination of a HaloTag on the protein of interest and a transiently binding fluorescent ligand opens up the possibility to do PAINT on both live and fixed samples. (B) By directly fusing a fluorophore to a transiently binding molecule, that is in turn binding to a peptide fused to the protein of interest, fixation compatible peptide-PAINT can be performed. Schematics created with Biorender.com. | ||
A multitude of fluorogenic approaches exist, these fluorogenic probes do not emit photons in solution, but only upon their interaction with their partner, greatly reducing the signal to noise ratio. Fluorescence-Activating and absorption-Shifting Tag (FAST) is one strategy that incorporates this principle by fusing the small Photoactive Yellow Protein (PYP) derived protein (14 kDa), a fluorogenic partner, to a variety of proteins of interest (POIs).44–47 A multitude of variants of FAST followed.48–50 However, so far due to low localization densities and the highly dynamic structures in live cells, reconstructions of nanoscale structures were difficult because long acquisition times were needed.47 More recently, another fluorogenic partner was reported, dye in Blc (DiB). This is based on the Blc protein, derived from E. coli, that was optimized by in silico mutagenesis for its fluorogen-activating properties. Upon binding of M739, a locked analogue of the GFP chromophore, the complex becomes fluorescent and as the interactions are weak, the interaction is reversible. This results in a combination of a highly stable localization density and high brightness, which makes DiB a promising marker in nanoscopy.51–53 More recently, transient interactions of fluorogenic rhodamine variants with the widely used HaloTags are reported as a versatile approach for PAINT. By modifying the ligand to bind with nanomolar dissociation constants, Kompa and colleagues reported transient interaction with two orthogonal variants of HaloTag7 that can be fused to a target protein of interest, which is illustrated in Fig. 5(A). This allows super-resolution imaging by PAINT, and by other super-resolution methods such as stimulated emission depletion (STED) microscopy MINFLUX.54 The major advantage of these modified ligands coupled to fluorogenic molecules is that they are highly robust and work well in live cells because they are membrane permeable, but the interaction is also maintained after cellular fixation. Furthermore, many proteins were already successfully fused and studied using the HaloTag, making the transiently binding ligands highly applicable in many multiplexed imaging applications for biological questions.
Finally, peptide–protein or protein–protein interactions can be utilized for PAINT. Eklund and colleagues first implemented this principle by replacing the DNA-docking strand on secondary antibodies for an alfa-helical docking-peptide that transiently interacted in a coiled–coil configuration with the imaging peptide coupled to a fluorophore. Because these coiled–coil interactions are highly tuneable by varying the repeat length, this approach resulted in fast, high quality reconstructions of the vimentin and microtubule network after antibody-docking staining and was termed peptide-PAINT.55 However, direct fusion of this docking-peptide proteins of interest, followed by imaging in fixed cells, may require caution as the lysines driving these coil–coil interactions can react with paraformaldehyde or glutaraldehyde affecting the interaction. Nevertheless, implementation of this interaction in fusion protein-based peptide-PAINT was recently shown by another study that used these tuneable interactions to perform fixed or live peptide-PAINT of membranous, cytoskeletal components, histones and receptors in epithelial cells and neurons. However, it should be noted that imaging of some targets in fixed cells required co-expression of docking peptides fused to endogenous binders, or antigen retrieval steps after fixation.56
Similar coil–coil interactions were also used for paint in live cells (Live-PAINT). Oi and colleagues expressed a number of proteins fused to one alfa helix of the heterodimeric SYNZIP pair together with the complementary helix fused to mNeonGreen in live S. cerevisiae. The transient interactions between these coils were sufficient for single molecule detection and tracking of several proteins of interest with diffraction unlimited resolution. It should be noted though that since this technique relies on the expression of the imaging peptide in live cells, the number of localizations is finite in theory as the expressed pool can be bleached and phototoxicity needs to be considered during the imaging.27
In addition to coiled-coils, we optimized the well-characterized interaction of the PDZ domain with a small sequence-optimized peptide for fusion protein-based peptide-PAINT. For this system, fusion of proteins of interest to the small peptide tag enables transient single molecule detection with purified PDZ domains coupled to a fluorophore after fixation. This interaction allowed for quantitative PAINT on nanoparticles and the interaction does not appear perturbed after fixation with both paraformaldehyde or glutaraldehyde as shown by imaging and reconstructions of both membranous (mitochondria) and cytoskeletal (vimentin) targets. This makes this interaction pair a good candidate for quantitative-PAINT and reconstruction in fixed cells expressing the peptide to a protein of interest. However, as the affinity of the PDZ domain is slightly lower then typically used for PAINT, more probe is required in solution, resulting in increased background signal during the acquisition. In addition to PAINT, we showed that the high affinity PDZclamp could be used to label the peptide-fused proteins for STED microscopy and, nanobodies could be fused to the peptide for more conventional staining-based PAINT experiments.26
For using peptide-PAINT, the peptide is usually chemically modified to become fluorescent, by direct labelling. Unfortunately, these dyes are usually charged, hydrophobic or induce non-specific binding. Using a fluorescent unnatural amino acid, these limitations can be overcome. Benzodiazole amino acids are suitable for this application, as they are optically tuneable by one atom replacement and they are compatible with solid-phase peptide synthesis, allowing for a generic labelling of protein sequences, whilst still retaining the structure of the non-labelled peptide. Furthermore, as they are fluorogenic, they are suitable for imaging with low background. The potential of these fluorogenic unnatural amino acids was shown by incorporating it in an engineered peptide that binds transiently to one of the PDZ domains of a postsynaptic density protein-95 (PSD-95), which is an important protein in the brain (Fig. 5(B)). This resulted in high-resolution molecular density maps of the PSD-95 protein distribution. Because these probes are small and easy to manufacture, they might be useful in minimally invasive and targeted biological imaging.57
Most of the mentioned fusion-proteins have excellent characteristics for live-cell imaging, as the probe is directly expressed in situ, sometimes with the need for the addition of a membrane-permeable fluorescent or fluorogenic molecule. In the future, we envision that fusion proteins will be further utilized for intracellular SMLM.
Synthetic probes
Originally, PAINT started with Nile red, a small molecule sensitive to the hydrophobic environment, to provide transient binding to lipid bilayers.58 Since then, synthetic probes have been explored as PAINT probes to go beyond samples that are normally not applicable to known fluorescent tags, especially those that cannot be labeled with targets. Due to their environment sensitivity, so far, these probes have proven more suitable in materials sciences than the probes mentioned before as they are not sensitive enough to single targets without defined uniform properties, such as specific proteins, ligands and lipids.Aloi and colleagues59 developed interfacial-PAINT (iPAINT) a polymer-based method to image all types of interfaces (solid–liquid, liquid–liquid and gas–liquid), a schematic representation is provided in Fig. 6(A). iPAINT consists of a large reservoir of synthesized polyethylene glycol (PEG) as a polymeric linker with end-functionalized photo-activable rhodamine dye (Cage552) for reversible, non-specific adsorption onto a wide range of interfaces. The dye functionalized PEG probes were UV-light activated and subsequently excited by a visible laser, for individual localization at the interface. Single molecule localization with a high signal to background ratio can be obtained since freely diffusing probes move much too fast for the EMCCD camera to detect. The synthesized probes thus acted as a model to achieve non-invasive, non-covalent super-resolution imaging of highly deformable interfaces.60 Another class of synthetic probes has been used in super-resolution imaging of supramolecular assemblies. These materials are composed of small building blocks connected through non-covalent bonds that are often weak making monomer–assembly interaction reversible and therefore useable for PAINT. The mechanical and chemical properties of supramolecular assemblies are strongly dependent on their structure, such as persistence length, type and number of crosslinks and geometry. Confocal and STED microscopy have been used for hydrogel imaging based on pre-labeled hydrogelators, and is therefore potentially an invasive approach and hinders the imaging of hydrogels in their native state.61 In that regard, it is advantageous to have a self-assembly building block labeled with a fluorescent dye and use the intrinsic assembly or binding dynamics of the same material for non-invasive in situ super-resolution imaging. Our group62 presented a PAINT approach on dipeptide hydrogels assembled with diphenylalanine protected at the N-terminus with the fluorenyl methoxycarbonyl group (FmocFF), where a Cy5-labeled FF was used as a PAINT probe to bind and unbind to FF assemblies reversibly (Fig. 6(B)). Due to the PAINT approach, a FmocFF hydrogel network could be imaged in 2D and 3D in its native conditions with a resolution down to ∼10 nm. The synthesized Cy5FF probe has thus extended the use of PAINT probes to native hydrogels.62
 | ||
| Fig. 6 Schematic examples of synthetic probes. (A) iPAINT uses polyethylene glycol (PEG) to characterize interfaces by non-invasive binding. (B) FmocFF and Thioflavin T use a similar principle, in which a single component of a dynamic system is fluorescently labeled and used for imaging of the dynamics and characteristics of a material. Schematics created with Biorender.com. | ||
Thioflavin T, an amyloidophilic dye used for staining of amyloid plaques found in for example Alzheimer's disease, is a synthetic molecule that not only binds transiently, but also increases fluorescence upon binding (Fig. 6(B)). The main advantage of using this probe is that it is not fluorescent in the unbound state, resulting in reconstructions with low background. This probe is applicable to a wide variety of amyloid structures, and it can even monitor the early formation of aggregates, which might be interesting for drug development. It is important to note that these aggregates were imaged on a coverslip and not in their natural environment.63 Recently, thioflavin X, a derivative of thioflavin T, was used to image the eccentricity of α-synuclein aggregates.64 These molecules were also further engineered to result in brighter molecules. ProteoStat and AT630 are two new variants of amyloidophilic dyes, which might label distinct features within these aggregates. AT630 is suitable for live cell imaging, which enables diagnosis in real-life samples especially at the early stages of the disease.65
Synthetic probes are so far mostly used for materials science, but they do also have potential in more biologically relevant samples, as described for the Thioflavin T probe. We envision that in the future, more and more synthetic probes will be developed.
Perspectives
Here we provided an overview of the wide variety of probes that have been developed for DNA-independent PAINT in order to localize, quantify and characterize specific targets with nanometric resolution. The main advantages of these probes lie within their ability to directly label targets of interest control the labelling stoichiometry or provide direct information about endogenous interactions and dynamics. Conversely, as many of these probes are based on highly specific and transient interactions their identification, benchmarking and tuning remain challenging and could be optimized in the future. We envision that complementary to DNA-PAINT, these probes will aid in obtaining a deeper understanding of both material development and biological phenomena. In an ideal world, a large toolbox of well-characterized and standardized sets probes can be combined to resolve cellular structures and materials of interest for multiple characteristics, in a high throughput fashion. Additionally, fusion-based and endogenous probes have the potential to provide more quantitative insights into protein numbers as well as into real biological interactions between cells and their environment, while synthetic probes might be more suitable for optimizing production of materials.For targets that do not have a probe with suitable kinetics, artificial intelligence (AI) powered probe design might pave the way to tailored PAINT probe design.66 Machine learning is predicted to revolutionize the protein engineering field, with AlphaFold now taking the lead. The method is able to accurately predict the structure of a protein based on their sequence. Language modeling algorithms such as ProGen have shown to be able to generate artificial proteins, with similar characteristics as natural proteins, but with a low sequence homology. By training the model with existing protein databases, the model learned to predict what the probability of the next amino acid in a sequence is, much like a language model that learns semantic and grammatical rules of a language.67 While at the moment it is not the aim of the model to predict proteins with a lower affinity than a natural protein, in the future it might be an interesting approach to find non-natural proteins or mutations in existing proteins that have an affinity within the PAINT regime. Additionally, with this method problems concerning live and fixed cell imaging could be tackled.
Conclusions
Over years of development in SRM techniques, DNA-PAINT has been shown to be one of the best performing SMLM tools due to its high specificity, high programmability, and multiplexing capability. While DNA-PAINT continuously proved to be a golden standard, non-DNA based PAINT probes have expanded the PAINT probe library to a wider range of samples, particularly in live cells and native synthetic materials where DNA-PAINT is limited. In this perspective we have reviewed four different types of emerging PAINT probes ranging from endogenous ligands for live cell SRM to synthetic small molecules for various materials. Nevertheless, several new PAINT methods beyond DNA have emerged which have experimentally solved the design puzzle and shown profound impact in expanding PAINT as a more general SRM technique. We envision that the various categories of PAINT probes we have covered in this paper will continue their momentum to improve our understanding of cell biology and to guide the design and synthesis of future soft matter materials in the biosensing, nanomedicine and food science industry.Author contributions
M. M. E. T. wrote the sections Introduction, Challenges for probe design, Engineered probes and compiled the review. R. P. T. wrote the sections Endogenous probes and Fusion-based probes. Y. W. and M. M. E. T. wrote the section Synthetic probes. Y. W. wrote the section Conclusion. M. M. E. T., R. P. T. and Y. W. revised the manuscript together. L. A. directed the writing of the review and contributed to all sections of the manuscript. All authors conceived the theme of the review and contributed to decisions on the overall content and the editing of the review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Dutch Research Council, through VIDI grant 192.028 to L. A. and M. M. E. T. and VENI grant 202.220 to R. P. T. All schematics were created using Biorender.com.Notes and references
- K. Xu, G. Zhong and X. Zhuang, Science, 2013, 339, 452–456 CrossRef CAS PubMed.
- R. Riera, J. Tauler, N. Feiner-Gracia, S. Borrós, C. Fornaguera and L. Albertazzi, ChemMedChem, 2022, 17(13), e202100633, DOI:10.1002/cmdc.202100633.
- C. V. Rimoli, C. A. Valades-Cruz, V. Curcio, M. Mavrakis and S. Brasselet, Nat. Commun., 2022, 13, 1–13 Search PubMed.
- L. Albertazzi, D. Van Der Zwaag, C. M. A. Leenders, R. Fitzner, R. W. Van Der Hofstad and E. W. Meijer, Science, 2014, 344, 491–495 CrossRef CAS PubMed.
- L. Schermelleh, A. Ferrand, T. Huser, C. Eggeling, M. Sauer, O. Biehlmaier and G. P. C. Drummen, Nat. Cell Biol., 2019, 21, 72–84 CrossRef CAS PubMed.
- S. Pujals, N. Feiner-Gracia, P. Delcanale, I. Voets and L. Albertazzi, Nat. Rev. Chem., 2019, 3, 68–84 CrossRef.
- M. Yamanaka, N. I. Smith and K. Fujita, Microscopy, 2014, 63, 177–192 CrossRef PubMed.
- S. Dhiman, T. Andrian, B. Santiago Gonzalez, M. M. E. Tholen, Y. Wang and L. Albertazzi, Chem. Sci., 2022, 13(8), 2152–2166, 10.1039/d1sc05506b.
- S. Samanta, W. Gong, W. Li, A. Sharma, I. Shim, W. Zhang, P. Das, W. Pan, L. Liu, Z. Yang, J. Qu and J. S. Kim, Coord. Chem. Rev., 2019, 380, 17–34 CrossRef CAS.
- J. J. A. Poole and L. B. Mostaço-guidolin, Cells, 2021, 10(7), 1760, DOI:10.3390/cells10071760.
- J. Valli, A. Garcia-Burgos, L. M. Rooney, B. V. de Melo e Oliveira, R. R. Duncan and C. Rickman, J. Biol. Chem., 2021, 297, 100791 CrossRef CAS PubMed.
- R. Jungmann, C. Steinhauer, M. Scheible, A. Kuzyk, P. Tinnefeld and F. C. Simmel, Nano Lett., 2010, 10, 4756–4761 CrossRef CAS PubMed.
- G. Jacquemet, A. F. Carisey, H. Hamidi, R. Henriques and C. Leterrier, J. Cell Sci., 2020, 133(11), jcs240713, DOI:10.1242/JCS.240713/VIDEO-3.
- A. Sharonov and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18911–18916 CrossRef CAS PubMed.
- O. K. Wade, J. B. Woehrstein, P. C. Nickels, S. Strauss, F. Stehr, J. Stein, F. Schueder, M. T. Strauss, M. Ganji, J. Schnitzbauer, H. Grabmayr, P. Yin, P. Schwille and R. Jungmann, Nano Lett., 2019, 19, 2641–2646 CrossRef CAS PubMed.
- S. Strauss and R. Jungmann, Nat. Methods, 2020, 17(8), 789–791, DOI:10.1038/s41592-020-0869-x.
- R. Jungmann, M. S. Avendaño, M. Dai, J. B. Woehrstein, S. S. Agasti, Z. Feiger, A. Rodal and P. Yin, Nat. Methods, 2016, 13(5), 439–442 CrossRef CAS PubMed.
- A. H. Clowsley, W. T. Kaufhold, T. Lutz, A. Meletiou, L. Di Michele and C. Soeller, Nat. Commun., 2021, 12, 1–10 CrossRef PubMed.
- R. van Wee, M. Filius and C. Joo, Trends Biochem. Sci., 2021, 46, 918–930 CrossRef CAS PubMed.
- J. Schnitzbauer, M. T. Strauss, T. Schlichthaerle, F. Schueder and R. Jungmann, Nat. Publ. Gr., 2017, 12(6), 1198–1228, DOI:10.1038/nprot.2017.024.
- T. Ha and P. Tinnefeld, Annu. Rev. Phys. Chem., 2012, 63, 595–617, DOI:10.1146/annurev-physchem-032210-103340.
- Q. Zheng, S. Jockusch, Z. Zhou and S. C. Blanchard, Photochem. Photobiol., 2014, 90, 448–454 CrossRef CAS PubMed.
- J. B. Grimm, A. K. Muthusamy, Y. Liang, T. A. Brown, W. C. Lemon, R. Patel, R. Lu, J. J. Macklin, P. J. Keller, N. Ji and L. D. Lavis, Nat. Methods, 2017, 14, 987–994 CrossRef CAS PubMed.
- M. V. Farrell, A. C. Nunez, Z. Yang, P. Pérez-Ferreros, K. Gaus and J. Goyette, Sci. Signaling, 2022, 15, eabg9782 CrossRef CAS PubMed.
- R. P. Tas, T. G. A. A. Bos and L. C. Kapitein, Methods Mol. Biol., 2018, 1665, 155–171 CrossRef CAS PubMed.
- R. P. Tas, L. Albertazzi and I. K. Voets, Nano Lett., 2021, 21, 9509–9516 CrossRef CAS PubMed.
- C. Oi, Z. Gidden, L. Holyoake, O. Kantelberg, S. Mochrie, M. H. Horrocks and L. Regan, Commun. Biol., 2020, 3, 458 CrossRef CAS PubMed.
- T. Kiuchi, M. Higuchi, A. Takamura, M. Maruoka and N. Watanabe, Nat. Methods, 2015, 12, 743–746 CrossRef CAS PubMed.
- L. S. Fischer, T. Schlichthaerle, A. Chrostek-Grashoff and C. Grashoff, ChemBioChem, 2021, 22, 2872–2879 CrossRef CAS PubMed.
- R. P. Tas, A. Chazeau, B. M. C. Cloin, M. L. A. Lambers, C. C. Hoogenraad and L. C. Kapitein, Neuron, 2017, 96, 1264–1271.e5 CrossRef CAS PubMed.
- D. Albrecht, C. M. Winterflood and H. Ewers, Methods Appl. Fluoresc., 2015, 3(2), 024001, DOI:10.1088/2050-6120/3/2/024001.
- S. Sograte-Idrissi, N. Oleksiievets, S. Isbaner, M. Eggert-Martinez, J. Enderlein, R. Tsukanov and F. Opazo, Cells, 2019, 8(1), 48, DOI:10.3390/cells8010048.
- G. Giannone, E. Hosy, F. Levet, A. Constals, K. Schulze, A. I. Sobolevsky, M. P. Rosconi, E. Gouaux, R. Tampe, D. Choquet and L. Cognet, Biophys. J., 2010, 99, 1303–1310 CrossRef CAS PubMed.
- R. Riera, T. P. Hogervorst, W. Doelman, Y. Ni, S. Pujals, E. Bolli, J. D. C. Codée, S. I. van Kasteren and L. Albertazzi, Nat. Chem. Biol., 2021, 17, 1281–1288 CrossRef CAS PubMed.
- S. Strauss, P. C. Nickels, M. T. Strauss, V. Jimenez Sabinina, J. Ellenberg, J. D. Carter, S. Gupta, N. Janjic and R. Jungmann, Nat. Methods, 2018, 15, 685–688 CrossRef CAS PubMed.
- P. Delcanale, D. Porciani, S. Pujals, A. Jurkevich, A. Chetrusca, K. D. Tawiah, D. H. Burke and L. Albertazzi, Angew. Chem., Int. Ed., 2020, 59, 18546–18555 CrossRef CAS PubMed.
- M. Sunbul, J. Lackner, A. Martin, D. Englert, B. Hacene, F. Grün, K. Nienhaus, G. U. Nienhaus and A. Jäschke, Nat. Biotechnol., 2021, 39, 686–690 CrossRef CAS PubMed.
- R. Jungmann, M. S. Avendaño, J. B. Woehrstein, M. Dai, W. M. Shih and P. Yin, Nat. Methods, 2014, 11, 313–318 CrossRef CAS PubMed.
- T. Schlichthaerle, M. Ganji, A. Auer, O. Kimbu Wade and R. Jungmann, ChemBioChem, 2019, 20, 1032–1038 CrossRef CAS PubMed.
- H. Gunasekara, R. Munaweera and Y. S. Hu, ACS Nano, 2022, 16, 129–139 CrossRef CAS PubMed.
- T. Miyoshi, Q. Zhang, T. Miyake, S. Watanabe, H. Ohnishi, J. Chen, H. D. Vishwasrao, O. Chakraborty, I. A. Belyantseva, B. J. Perrin, H. Shroff, T. B. Friedman, K. Omori and N. Watanabe, Cell Rep., 2021, 34, 108708 CrossRef CAS PubMed.
- T. Miyoshi, T. B. Friedman and N. Watanabe, STAR Protoc., 2021, 2, 100967 CrossRef CAS PubMed.
- Q. Zhang, A. Miyamoto, S. Watanabe, T. Arimori, M. Sakai, M. Tomisaki, T. Kiuchi, J. Takagi and N. Watanabe, Cells Rep. Methods, 2022, 100301 CrossRef CAS PubMed.
- M. A. Plamont, E. Billon-Denis, S. Maurin, C. Gauron, F. M. Pimenta, C. G. Specht, J. Shi, J. Quérard, B. Pan, J. Rossignol, N. Morellet, M. Volovitch, E. Lescop, Y. Chen, A. Triller, S. Vriz, T. Le Saux, L. Jullien and A. Gautier, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 497–502 CrossRef CAS PubMed.
- C. Li, M.-A. Plamont, H. L. Sladitschek, V. Rodrigues, I. Aujard, P. Neveu, T. Le Saux, L. Jullien and A. Gautier, Chem. Sci., 2017, 8(8), 5598–5605, 10.1039/c7sc01364g.
- H. Benaissa, K. Ounoughi, I. Aujard, E. Fischer, R. Goïame, J. Nguyen, A. G. Tebo, C. Li, T. Le Saux, G. Bertolin, M. Tramier, L. Danglot, N. Pietrancosta, X. Morin, L. Jullien and A. Gautier, Nat. Commun., 2021, 12, 1–17 Search PubMed.
- E. M. Smith, A. Gautier and E. M. Puchner, ACS Chem. Biol., 2019, 14, 1115–1120 CrossRef CAS PubMed.
- A. G. Tebo and A. Gautier, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- A. G. Tebo, B. Moeyaert, M. Thauvin, I. Carlon-Andres, D. Böken, M. Volovitch, S. Padilla-Parra, P. Dedecker, S. Vriz and A. Gautier, Nat. Chem. Biol., 2021, 17(1), 30–38, DOI:10.1038/s41589-020-0611-0.
- K. S. Mineev, S. A. Goncharuk, M. V. Goncharuk, N. V. Povarova, A. I. Sokolov, N. S. Baleeva, A. Y. Smirnov, I. N. Myasnyanko, D. A. Ruchkin, S. Bukhdruker, A. Remeeva, A. Mishin, V. Borshchevskiy, V. Gordeliy, A. S. Arseniev, D. A. Gorbachev, A. S. Gavrikov, A. S. Mishin and M. S. Baranov, Chem. Sci., 2021, 12, 6719–6725 RSC.
- N. G. Bozhanova, M. S. Baranov, N. V. Klementieva, K. S. Sarkisyan, A. S. Gavrikov, I. V. Yampolsky, E. V. Zagaynova, S. A. Lukyanov, K. A. Lukyanov and A. S. Mishin, Chem. Sci., 2017, 8, 7138–7142 RSC.
- N. G. Bozhanova, A. S. Gavrikov, A. S. Mishin and J. Meiler, Sci. Rep., 2020, 10, 11049 CrossRef CAS PubMed.
- L. Muslinkina, A. S. Gavrikov, N. G. Bozhanova, A. S. Mishin, M. S. Baranov, J. Meiler, N. V. Pletneva, V. Z. Pletnev and S. Pletnev, ACS Chem. Biol., 2020, 15, 2456–2465 CrossRef CAS PubMed.
- J. Kompa, J. Bruins, M. Glogger, J. Wilhelm, M. S. Frei, M. Tarnawski, E. D’Este, M. Heilemann, J. Hiblot and K. Johnsson, bioRxiv, 2022, 2022.06.20.496706.
- A. S. Eklund, M. Ganji, G. Gavins, O. Seitz and R. Jungmann, Nano Lett., 2020, 20, 6732–6737 CrossRef CAS PubMed.
- B. K. Maity, D. Nall, Y. Lee and P. R. Selvin, Small Methods, 2022, 7(4), 2201181, DOI:10.1002/smtd.202201181.
- A. F. De Moliner, Z. Konieczna, L. Mendive, R. Saleeb, K. Morris, J. A. Gonzalez, T. Kaizuka, S. Grant and M. Horrocks, Angew. Chem., Int. Ed., 2023, 62(4), e202216231, DOI:10.1002/anie.202216231.
- A. Sharonov and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 18911–18916 CrossRef CAS PubMed.
- A. Aloi, N. Vilanova, L. Albertazzi and I. K. Voets, Nanoscale, 2016, 8, 8712–8716 RSC.
- B. Adelizzi, A. Aloi, N. J. Van Zee, A. R. A. Palmans, E. W. Meijer and I. K. Voets, ACS Nano, 2018, 12, 4431–4439 CrossRef CAS PubMed.
- R. Kubota, K. Nakamura, S. Torigoe and I. Hamachi, ChemistryOpen, 2020, 9, 67–79 CrossRef CAS PubMed.
- E. Fuentes, K. Boháčová, A. M. Fuentes-Caparrós, R. Schweins, E. R. Draper, D. J. Adams, S. Pujals and L. Albertazzi, Chem. – Eur. J., 2020, 26, 9869–9873 CrossRef CAS PubMed.
- K. Spehar, T. Ding, Y. Sun, N. Kedia, J. Lu, G. R. Nahass, M. D. Lew and J. Bieschke, ChemBioChem, 2018, 19, 1944–1948 CrossRef CAS PubMed.
- D. Emin, Y. P. Zhang, E. Lobanova, A. Miller, X. Li, Z. Xia, H. Dakin, D. I. Sideris, J. Y. L. Lam, R. T. Ranasinghe, A. Kouli, Y. Zhao, S. De, T. P. J. Knowles, M. Vendruscolo, F. S. Ruggeri, F. I. Aigbirhio, C. H. Williams-Gray and D. Klenerman, Nat. Commun., 2022, 13, 1–15 Search PubMed.
- M. J. Morten, L. Sirvio, H. Rupawala, E. M. Hayes, A. Franco, C. Radulescu, L. Ying, S. J. Barnes, A. Muga and Y. Ye, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, 1–12 CrossRef PubMed.
- M. Eisenstein, Nat. Biotechnol., 2023, 41, 303–305 CrossRef CAS PubMed.
- A. Madani, B. Krause, E. R. Greene, S. Subramanian, B. P. Mohr, J. M. Holton, J. L. Olmos, C. Xiong, Z. Z. Sun, R. Socher, J. S. Fraser and N. Naik, Nat. Biotechnol., 2023, 2023, 1–8 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |