
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLab-on-a-DNA origami: nanoengineered single-molecule platforms
Sergio
Kogikoski
Jr
 ,
João
Ameixa
,
Amr
Mostafa
and
Ilko
Bald
*
,
João
Ameixa
,
Amr
Mostafa
and
Ilko
Bald
*
Institute of Chemistry, Hybrid Nanostructures, University of Potsdam, Karl-Liebknecht-Str. 24-25, 14476, Potsdam, Germany. E-mail: bald@uni-potsdam.de
First published on 31st March 2023
Abstract
DNA origami nanostructures are self-assembled into almost arbitrary two- and three-dimensional shapes from a long, single-stranded viral scaffold strand and a set of short artificial oligonucleotides. Each DNA strand can be functionalized individually using well-established DNA chemistry, representing addressable sites that allow for the nanometre precise placement of various chemical entities such as proteins, molecular chromophores, nanoparticles, or simply DNA motifs. By means of microscopic and spectroscopic techniques, these entities can be visualized or detected, and either their mutual interaction or their interaction with external stimuli such as radiation can be studied. This gives rise to the Lab-on-a-DNA origami approach, which is introduced in this Feature Article, and the state-of-the-art is summarized with a focus on light-harvesting nanoantennas and DNA platforms for single-molecule analysis either by optical spectroscopy or atomic force microscopy (AFM). Light-harvesting antennas can be generated by the precise arrangement of chromophores to channel and direct excitation energy. At the same time, plasmonic nanoparticles represent a complementary approach to focus light on the nanoscale. Plasmonic nanoantennas also allow for the observation of single molecules either by Raman scattering or fluorescence spectroscopy and DNA origami platforms provide unique opportunities to arrange nanoparticles and molecules to be studied. Finally, the analysis of single DNA motifs by AFM allows for an investigation of radiation-induced processes in DNA with unprecedented detail and accuracy.
 Sergio Kogikoski | Dr Sergio Kogikoski Jr. received his BSc in Science and Technology from the Federal University of ABC (UFABC, Brazil) and his PhD in Chemistry from the Federal University of ABC (UFABC, Brazil). After a postdoctoral period at the State University of Campinas (UNICAMP, Brazil) studying DNA electrochemical properties, he has worked since 2019 as a postdoctoral researcher in Prof. Ilko Bald's group at the University of Potsdam (UP, Germany). His work nowadays focuses on using plasmonic nanostructures to study plasmon-induced processes, principally using light-generated hot-charge carriers in chemical reactions. |
 João Ameixa | João Ameixa obtained a MSc degree in Biomedical Engineering at the University NOVA of Lisbon (PT). In 2020, he received a PhD degree in Physics at the University of Innsbruck (AT) as well as in Radiation Biology and Biochemistry – Applied Atomic and Molecular Physics at the University NOVA of Lisbon (PT) through a co-tutelle agreement between both institutions. Since 2021, he is working as a post-doctoral researcher in the Hybrid Nanostructures’ group led by Prof. Dr Ilko Bald at the University of Potsdam (DE). He currently uses DNA origami nanostructures in combination with atomic force microscopy to investigate radiation damage to DNA. |
 Amr Mostafa | Amr Mostafa received his BSc in Pharmacy and Biotechnology from the German University in Cairo (GUC) and his MSc in Pharmaceutical Biotechnology from Martin Luther University of Halle-Wittenberg, Germany. Since 2018 he has been a PhD student in Prof. Ilko Bald's group at the University of Potsdam. His work nowadays focuses on designing and using DNA origami nanostructures to create plasmonic nanostructures for the analysis of proteins by surface-enhanced Raman scattering (SERS). |
 Ilko Bald | Ilko Bald studied chemistry at the FU Berlin/D where he also obtained a PhD in Physical Chemistry. After a postdoc at the University of Iceland he moved to the Interdisciplinary Nanoscience Center (iNANO) at Aarhus University/DK working on scanning probe microscopy and DNA nanotechnology. In 2013 he established a junior research group “Optical Spectroscopy and Chemical Imaging” at the University of Potsdam and the Federal Institute for Materials Research and Testing (BAM, Berlin/D). Since 2019 he is Professor for Hybrid Nanostructures at the University of Potsdam investigating nanomaterials, their optical properties, and electron induced processes. |
1. Nanoscale assembly using DNA origami nanostructures
Miniaturization is a fundamental idea for the advancement of chemistry in the following decades since the potential benefits include improved sensitivity, increased reaction speed, reduced cost, and reduced waste. Lab-on-a-chip technologies are already available and combine in only one chip different chemical processes that allow the preparation, separation, detection, and analysis of molecules, principally in biomedical diagnosis. Nevertheless, advancements in chemistry and nanotechnology enable further miniaturization, and it is now possible to perform physical–chemical analysis at the single-molecule level. Here we show some of the progress enabled by DNA origami for studies in the few- to single-molecule regime. We envisage that the contributions shown here will further develop Lab-on-a-DNA origami techniques.DNA origami is a highly interdisciplinary research field, unifying concepts from chemistry, physics, biology, and engineering to further develop and explore the limits of DNA in creating unique nanostructures and exploiting them for new applications. DNA nanotechnology uses a bottom-up strategy to form nanostructures. Compared to other molecules, DNA has well-predictable interactions derived from the very specific Watson–Crick–Franklin base pairing model, giving rise to nanostructures programmed by the DNA sequence. DNA chemistry also enables the precise orthogonal placement of various chemical modifications. An excellent recent review by Seeman and Sleiman1 is suggested for those interested in the historical evolution of DNA nanotechnology.
Even though DNA nanotechnology started in the 1980s, the DNA origami technique was only developed in 2006, when the idea of using designed DNA strands to fold a single-stranded viral DNA into any desired shape was first shown by Paul Rothemund.2 From the first publication in 2006 until now, the field evolved from simple two-dimensional nanostructures to very complex 3D, dynamical, and fully functional assemblies for various technological applications.3 An interesting review by Tapio and Bald4 discusses many experimental strategies to obtain novel DNA origami nanostructures. At the same time, recent reviews focused on specific applications for DNA origami nanostructures, such as drug delivery,5 bioanalytical chemistry,6 plasmonics, and photonics.7 Single DNA origami structures can easily be analyzed by atomic force microscopy (AFM), which was the basis for early studies to use DNA origami as platforms for single-molecule placement and detection, such as RNA detection by AFM.8,9 Optical studies followed later using single-molecule fluorescence methods taking advantage of the precise placement of dyes with well-defined distances.10 For other optical techniques, such as Raman scattering, signal-enhancing elements, such as plasmonic nanoparticles, are required.
Herein we will present some developments to enable single-molecule physical–chemical studies using DNA origami and opportunities to explore in the future. In this direction, we give a short overview of the basics of DNA origami, the use of DNA programmability in generating light-harvesting nanoantennas in the few-molecules regime, and later the use of DNA origami as single molecule platforms for surface-enhanced spectroscopy studies and also as a method to directly quantify DNA radiation damage of DNA strands.
1.1. The concept of DNA origami nanostructures
DNA origami technology is a technique to create well-defined and almost arbitrarily shaped 2D and 3D nanostructures, which are programmed from DNA. The basic idea is to fold a long single-stranded DNA chain with the help of short single-stranded DNA staple strands. The design concepts of DNA nanotechnology are based on the B-type DNA structure, which has 0.34 nm of distance between two base pairs (bp), and every turn of the helix comprises about 10.5 bp (3.6 nm), giving rise to a diameter of the double helix of about 2 nm.11 With the structural information and design concepts available nowadays, it is possible to design any nano-object made entirely from DNA. The shape can be controlled by arranging DNA double helices in hexagonal or square cross sections, and curved structures can be obtained by systematically introducing mismatches.12,13 The size of a single DNA origami structure is limited by the length of the scaffold strand. Recently, a concept for tailor-made scaffolds has been introduced. Wireframe DNA origami structures are larger structures with lower DNA density and higher flexibility. Larger, micrometer-sized DNA structures can be assembled by assembling individual DNA origami building blocks into large-scale structures.14–18 For a recent and profound review of DNA origami technology, refer to Dey et al.19DNA origami presents two fundamental properties for single molecule studies: the orthogonal position placement of chemical entities on the DNA origami platform due to the well-known B-type DNA structure and the versatile possibilities for chemical functionalization. Both properties are nowadays easily achieved due to the advancements in the chemical synthesis of DNA. In this way, after the nanostructure design, enabled by many available open-source software packages, such as cadnano and others,20 it is possible to place chemical moieties, positioned with nanometer precision and study single molecule chemical processes with a large variety of experimental physical chemical techniques for this review more notably optical spectroscopy and AFM.
Some DNA origami shapes are well-established due to their simple preparation and high stability, such as Rothemund's triangle, nanoframes, 6-helix bundles, and DNA rectangles. Since most of those shapes are simple 1D or 2D nanostructures, it is easy to precisely modify distances between moieties, such as fluorophores or enzymes in enzymatic cascades. However, 3D shapes favor arranging optically active functionalities in three dimensions for spectroscopic studies. Moreover, 3D structures are less flexible than 2D structures, making it easier to implement dynamic structural changes. Some examples of DNA origami nanostructures already used as single-molecule platforms for chemical studies are presented in Fig. 1.
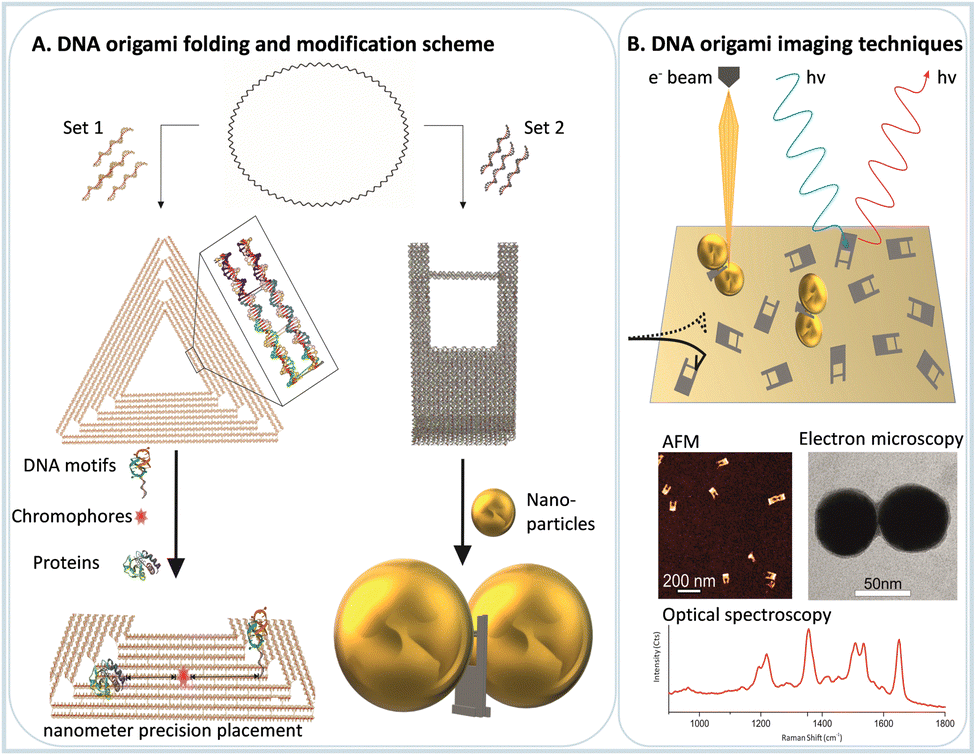 | ||
| Fig. 1 (A) DNA origami folding scheme. From a single stranded circular viral DNA two different sets of staple strands are mixed and after proper incubation two different nanostructures are formed, a 2D nanotriangle and a 3D nanofork. The nanotriangle is a perfect canvas to precisely place different functional groups with nanometer precision and it is usually a good starting point for later optimization. The nanofork is a 3D nanostructure used to self-assemble nanoparticle dimers for single molecule SERS. (B) Some of the techniques used to characterize physico-chemical processes on DNA origami platforms, and examples of AFM and transmission electron microscopy images, and a SERS spectrum of a single dimer. | ||
2. Light-harvesting nanoantennas
Light harvesting and energy transfer are the most important topics in science and engineering. This is due to the high relevance of future technologies that transform sunlight into electrical or chemical energy. In nature, light harvesting and energy transfer were optimized such that the energy pathways occur without significant losses. This is achieved by carefully positioning different proteins within the cellular environment, for example in the natural occurring photosynthetic protein complex. Being inspired by nature, there is a strong desire to create artificial light-harvesting systems for other purposes. However, conventional self-assembly processes make it very difficult to precisely organize different components in an ordered manner to achieve maximum response. In this sense, the orthogonal placement of various elements on DNA origami can be used to optimize many parameters to obtain the highest energy conversion efficiency. Herein we call such assemblies nanoantennas since they can also concentrate light. Below we will discuss two kinds of structures used to harvest light, the first based on chromophores usually studied by fluorescence spectroscopy, and the second using plasmonic metal nanoparticles, which are used both in surface-enhanced Raman scattering (SERS) and surface-enhanced fluorescence (SEF).2.1. Fluorescence-based nanoantennas
After the initial years of DNA origami research, when the field used the versatility of the technique to evolve from simple 2D nanostructures to complex and dynamical 3D creations, researchers started to use the many functionalities that such systems provide to study energy transfer processes.7 Due to the simple functionalization of DNA origami with organic dyes, early studies focused on fundamental Förster resonance energy transfer (FRET) processes. FRET refers to the dipole-mediated energy transfer between an excited donor dye and an acceptor dye. For FRET to occur, the fluorescence emission must overlap with the acceptor's absorption. Both chromophores must be at a close distance, i.e., typically between 1 nm and 10 nm, depending on the characteristic Förster radius R0 of the donor–acceptor pair, Fig. 2A. The following dependence on the distance between dyes gives the FRET efficiency: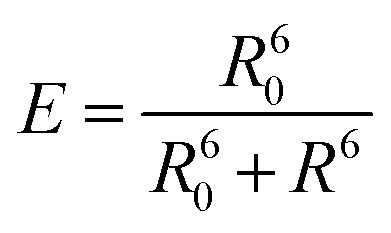 | (1) |
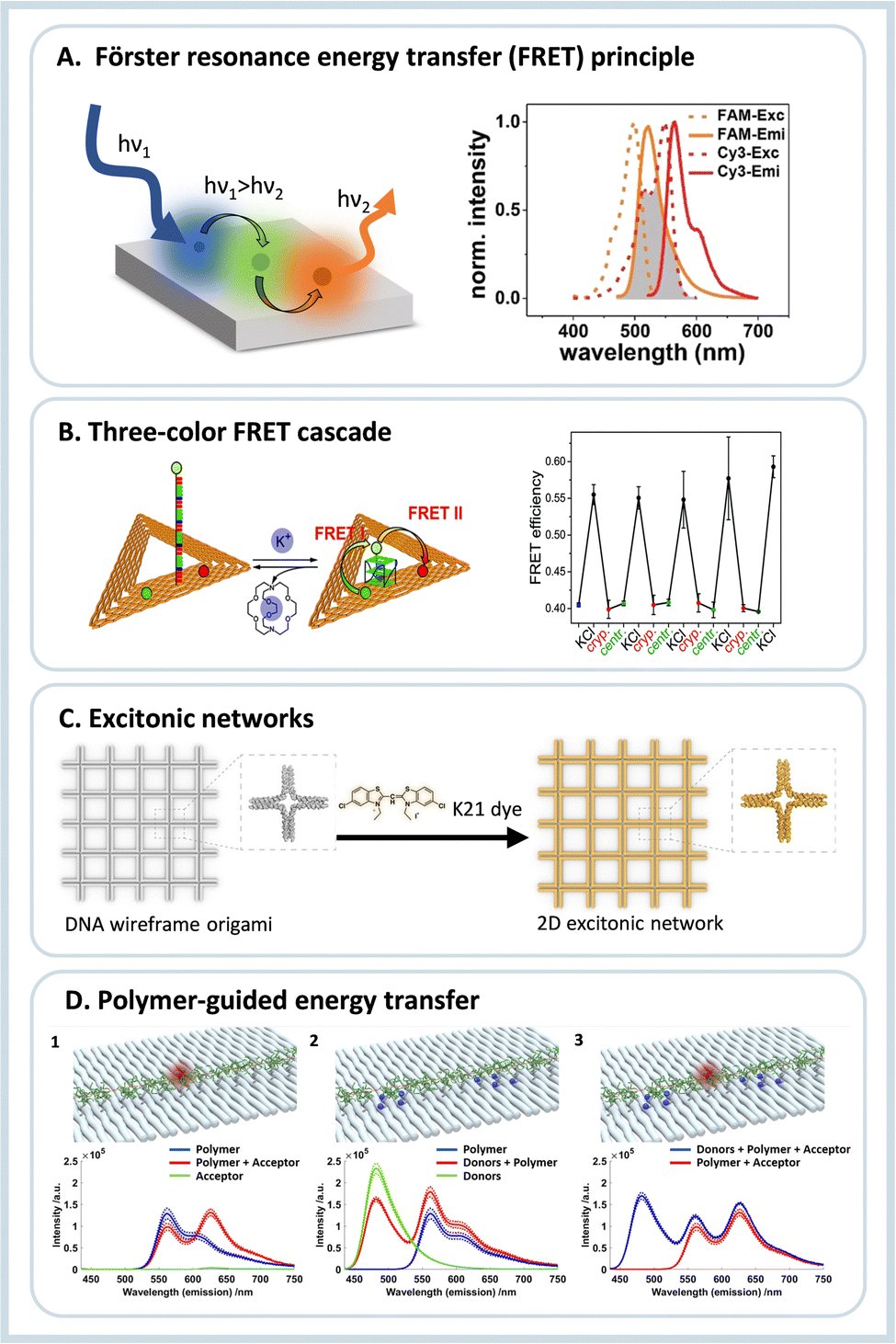 | ||
| Fig. 2 (A) Schematics of a FRET assembly containing three different dyes, on the right side the required spectral overlap is shown between the emission and absorption between two dyes able to transfer energy by FRET. (B) Three-color FRET cascade self-assembled using DNA origami nanotriangles, and the operational principle of the K+ sensor due to G-quadruplex formation, and on the right the switching of the FRET efficiency due to presence or absence of K+ ions is shown. Reproduced from ref. 21 with permission from Royal Society of Chemistry. (C) DNA origami wireframe that generates a 2D excitonic network upon incubation with a K21 dye, which can transfer energy multi-directionally. Reproduced from ref. 22 with permission from Wiley. (D) Illustration and fluorescence emission spectra for investigation of the individual energy transfer processes between a polymer and fluorophores when excited at 425 nm. (1) Assembly containing only the polymer and acceptor molecule; (2) assembly containing the polymer and the donor group; (3) full assembly containing the donor, polymer and acceptor. Reprinted with permission from ref. 23. Copyright 2023 American Chemical Society. | ||
It can be determined experimentally either from the fluorescence intensities of the donor alone (ID) and its intensity in the presence of the acceptor (IDA) or from the respective fluorescence decay times (τD and τDA, respectively):
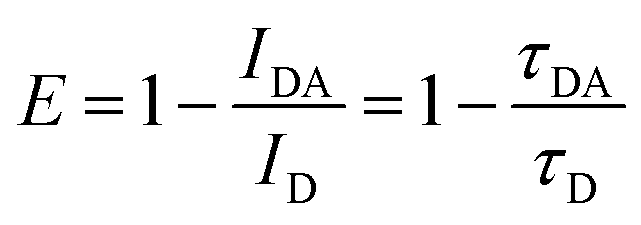 | (2) |
The group of Yan Liu used a 3D DNA origami structure to show that DNA could be used to channel the energy absorbed by ethylpyrene dyes (primary donor) to an intermediate donor (cyanine 3) and finally to an acceptor chromophore (Alexa Fluor 647) at the tip of the structure.24 Phillip Tinnefeld's group used a rectangular DNA origami block to study the distance dependence of FRET within the distance range between 2.5 and 14 nm.25 The same group studied directional energy transfer pathways at a single-molecular level, again using a blue fluorophore (ATTO488) as the input dye, ATTO565 as an energy conductor or jumper dye placed between two other dyes, the red ATTO647N and the IR fluorophore Alexa 750 as output acceptors.26 After those three seminal works, the topic blossomed.
After the initial works showing the capabilities to create assemblies to transfer energy, such systems were applied to analytical processes and single-molecule devices to study charge transfer processes in the single-molecule regime. DNA nanotriangles were equipped with telomeric DNA sequences that form G-quadruplexes in the presence of monocations.27 The G-quadruplex structure allows the assembly to act as a sensor. In the presence of potassium ions, the G-quadruplex closes, enabling the energy transfer between the dyes of a FRET chromophore pair, one attached to the DNA origami platform and the other attached to the 5′ end of the G-rich sequence extending from the DNA origami. Interestingly, the G-rich sequence alone (without the DNA origami platform) also folds into a G-quadruplex in the presence of Na+ ions. However, this is not the case when it is attached to the DNA origami structure. This effect is ascribed to steric hindrance and/or kinetic trapping and allows for selective sensing of K+ ions even in the presence of high concentrations of Na+. This work was later expanded to allow the fabrication of switchable photonic wires consisting of three and four-color FRET cascades, Fig. 2B.21 Also, due to the very high selectivity of the G-quadruplex towards potassium,28 the creation of more advanced photonic circuits or DNA-based logic gates is possible.
The conjugation of FRET pairs on DNA origami was further optimized to achieve maximum energy transfer efficiency by increasing the number of donors and acceptors and systematically varying their relative position.29 Structures containing the dyes were arranged in arrays of 3 × 4, with an average distance of 6.8 nm between dyes, obtaining FRET efficiencies in the order of 0.65. The optimized array was then used to obtain a ratiometric pH sensing system using a pH-responsive dye as an acceptor. Again, the design was optimized regarding its analytical capabilities and showed a linear behavior in the pH range between 5 and 8. This served as a proof-of-concept for a versatile ratiometric sensing scheme in which the acceptor dye can be easily replaced with another analyte-responsive dye.
Hemmig and co-authors also showed the optimization of a ring-like antenna. The maximum efficiency was achieved with one acceptor molecule in the center of a ring with six other donors.30 Our group studied a similar assembly, in which the stoichiometry between the donor and transmitter molecules was varied to obtain the maximum FRET efficiency and antenna effect AE, obtained as31
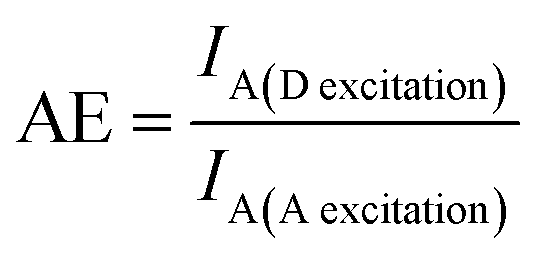 | (3) |
In this arrangement, AE (which is, in contrast to the FRET efficiency, an acceptor property) was larger than 1, i.e., the fluorescence intensity of the acceptor was higher upon donor excitation (IA(D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) excitation)) than direct acceptor excitation (IA(Aexcitation)). In general, the results showed that the FRET efficiency could be increased by tuning the spectral properties of the molecules. At the same time, the antenna effect is strongly influenced by the number of energy-delivering pathways directly related to the number of donor and transmitter molecules. In another work, energy transfer using the same kind of fluorophores (HOMO-FRET) was also tested and showed the possibility of transferring energy directionally over a distance of 16 nm.32
excitation)) than direct acceptor excitation (IA(Aexcitation)). In general, the results showed that the FRET efficiency could be increased by tuning the spectral properties of the molecules. At the same time, the antenna effect is strongly influenced by the number of energy-delivering pathways directly related to the number of donor and transmitter molecules. In another work, energy transfer using the same kind of fluorophores (HOMO-FRET) was also tested and showed the possibility of transferring energy directionally over a distance of 16 nm.32
More recently, DNA origami was used to create excitonic wires and networks.22,33 DNA origami allows precise control over the geometric arrangement and energy flow through photonic materials. The DNA double helix was used to guide the assembly of the cyanine dye K21, which forms a closed-packed aggregate exhibiting strong excitonic coupling between chromophores, Fig. 2C. In the first work, an excitonic wire was created from the DNA double helix with a maximum energy transfer distance of about 100 bp (∼32 nm) with donor-to-bridge-to-acceptor transfer efficiency in the order of 90%.34 After this, larger DNA origami nanowires were realized using 4-helix bundles achieving 500 nm energy transfer distance, and even more complex photonic assemblies were fabricated based on modular building blocks.33 The same strategy was used to create 2D wireframe DNA origami nanostructures for multiple excitation energy transfer pathways increasing the light collection efficiency, decreasing possible failures, and enabling a more robust and resilient system.22
Due to the relatively straightforward chemical functionalization of DNA, energy and charge transfer between short polymers were also already studied. For example, Wang et al.35 modified DNA frames with polyaniline and poly(phenylenevinylene), which would reversibly change a fluorescence signal output upon a redox reconfiguration. More recently, Madsen et al.23 modified a phenylene–vinylene-based polymer with multiple DNA strands to immobilize the polymer linearly extended in a DNA origami structure. Later donor and acceptor dyes were incorporated into DNA at various distances to study the energy transfer from the donor to the polymer to the acceptor, Fig. 2D. The energy transfer efficiency was calculated to be around 5% on the longest separation distance. Those works show the possibilities of using more complex structures on DNA origami nanostructures to better understand energy transfer processes in complex molecules and to create more advanced light-harvesting systems.
Energy transfer studies represent exciting uses of DNA nanostructures since it is easy to precisely modify the position, orientation, and other properties of the involved entities, mainly by changing a few DNA sequences. Moreover, we can observe that the topic is recently evolving from simple FRET pairs studies to more complex energy transfer pathways such as the excitonic networks and the use of conjugated polymers, opening many possibilities to use DNA in more sophisticated photonics studies. In the next section, the conjugation of nanoparticles to the DNA origami will be discussed, introducing another optically active entity that enables even more complex energy transfer processes.
2.2. Nanoantennas based on plasmonic nanoparticles
One advancement in the study of nanoantennas was the introduction of nanoparticles to DNA nanostructures, principally plasmonic nanoparticles. Plasmonic nanoparticles, in most cases made of gold and silver (Au and Ag), are versatile materials that resonantly absorb light in the visible region of the electromagnetic spectrum.36 After light absorption, a cascade of different processes is triggered, which can lead to the generation of hot charge carriers and later thermalization of the system.37 Due to the easy modification of metallic nanoparticles with DNA sequences containing thiolated ligands, it is possible to build many different constructs by self-assembly to study other light-harvesting processes.The first work showing an energy transfer process between a 10 nm AuNP and a fluorophore on DNA origami was published in 2012.38 Again, the ability of DNA to place the dye at specific distances was essential. The experiments showed that at any distance below 15 nm, the fluorescence emission was strongly quenched by the nanoparticle. Later a similar study was performed with a semiconductor quantum dot in proximity to a 30 nm AuNP. Again, a strong quenching of the fluorescence emission was observed. The study also showed that increased nonradiative decay pathways dominated the quenching effect with semiconductor quantum dots.39 Both works envisaged that such assemblies would be used as nano rulers, but it is a technology that has yet to be fully employed. At the same time, nano rulers based on DNA origami and chromophoric groups are already widely used in superresolution microscopy.40,41
A more advanced example used AuNPs to transfer the absorbed energy to a transmitter and an acceptor, measuring the output intensity in a PRET (plasmon-coupled resonance energy transfer) scheme.42 In this system, DNA origami organized the AuNPs and the dyes with a specific distance between chromophoric centers. The results showed that when the AuNP was excited, the FRET efficiency was enhanced 5-fold. The authors showed that the energy pathway in such a system is bidirectional, i.e., the nanoparticles act as an energy donor and an energy transmitter, opening multiple energy transfer pathways. Also, the electric field around the nanoparticles plays an essential role in enhancing fluorescence emission. Another work also tested FRET in the presence of 5 nm AuNP and showed only moderate enhancement of the FRET efficiency. However, at the same time, the work showed that this enhancement is mainly given by an increased nonradiative contribution to the transfer rate, thus increasing the FRET action radius in the presence of the AuNP.43 Those works show that the design of such materials is more complex and challenging compared to simple FRET systems. However, it could lead to the development of biomimetic energy harvesting systems.
Since the initial development of the DNA origami technique, several works have focused on the creation of assemblies containing multiple nanoparticles in a more straightforward methodology compared to lithography, for example.8,44 If it was possible to arrange plasmonic nanoparticles with small inter-particle gaps, such complex assemblies could eventually create plasmonic waveguides. The first work showing the use of DNA to create plasmonic waveguides was published in 2016 by Pirzer et al.45 In this work, DNA origami organized five 10 nm AuNPs linearly over about 60 nm. An acceptor dye was introduced on one end, and an emitter dye was added on the other. Due to the plasmonic coupling between the nanoparticles, the energy absorbed by one dye was transferred through the nanoparticle chain, and the emission of the different dye was observed. Also, a thermoresponsive peptide was added to the system. The signal varied up and down as the system was heated and cooled, and the energy transmission was only happening in the perfectly assembled waveguide, Fig. 3A.
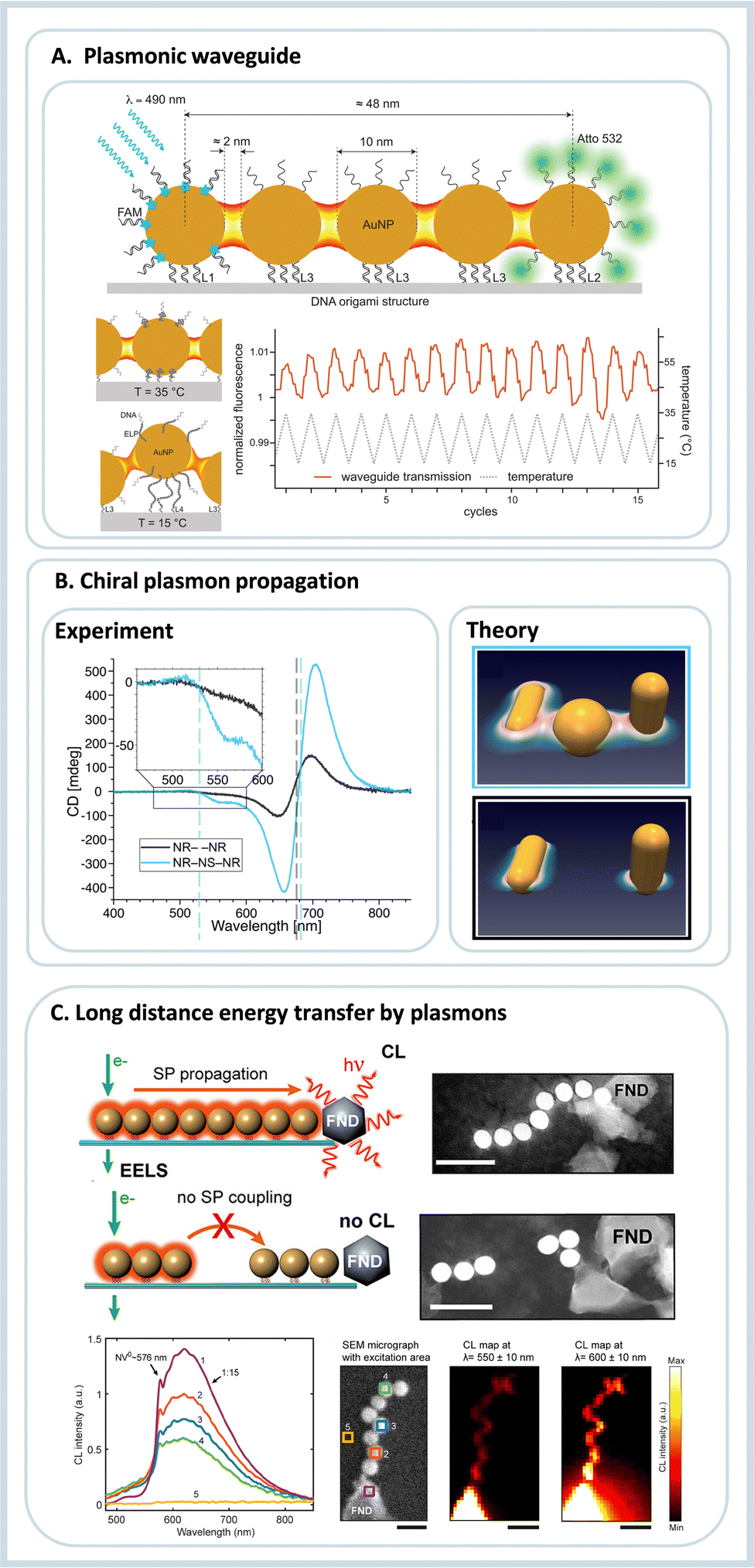 | ||
| Fig. 3 Energy transfer guided by plasmonic nanoparticles self-assembled using DNA origami. (A) Plasmonic waveguide containing five gold nanoparticles, at one end donor molecules are placed. The absorbed energy is transferred by the AuNP and later the energy is emitted by an acceptor. The same system shows reversible thermoresponsiveness enabling on and off states of energy transfer. Reproduced with permission from ref. 45. Copyright 2023 American Chemical Society. (B) Chiral plasmonic assembly using Au nanorods and a AuNP, due to the appropriate plasmon coupling the circular dichroism signal is enhanced compared to the incomplete assembly. Reproduced with permission from ref. 44. (C) Plasmonic waveguide showing energy waveguiding through AuNPs to a fluorescent nanodiamond. Below the cathodoluminescece emission dependent of the excitation placement is shown. Reproduced with permission from ref. 46. Copyright 2023 American Chemical Society. | ||
Later, Roller et al.47 assembled a trimeric structure containing two AuNPs interspaced by an AgNP, and the plasmonic properties of the construct were studied using scattering spectroscopy and computational simulations. The results showed a very efficient passage of the plasmon from one AuNP to the other through the AgNP. More than this, the process was suggested to be ultrafast and to happen without any significant energy loss at the transmitting element. A similar strategy was used to study chiral plasmonic transfer over long distances: Two gold nanorods (AuNR) were assembled using DNA origami in a Z-shape, Fig. 3B. This construct presented a chiral response to light, but the chiral response was amplified when a spherical gold nanoparticle was introduced in the interspace between the two nanorods. The results showed both experimentally and computationally that the bridging nanoparticle is increasing the signal due to the generated hot spot between NP and NR and also by the near-field around the plasmonic nanoparticles, Fig. 3B.44
Using a 6-helix DNA origami bundle, Gür et al.46 linearly arranged eight 40 nm AuNP as a waveguide.35 Due to the placement provided by DNA, the distance between the nanoparticles was in the order of 2 nm. This spacing reduced the energy propagation losses dramatically. The waveguide was later conjugated to a fluorescent nanodiamond at one end. The energy transfer was tested by electron energy-loss spectroscopy and cathodoluminescence imaging spectroscopy.46 Using microscopy methods, one end of the assembly was optically excited, and the fluorescence emission was observed on the other. When the waveguide was complete, the nanodiamond fluorescence was observed, and when one or two nanoparticles were missing in the waveguide, the fluorescence decreased dramatically. The same nanoparticle assembly was also coupled to silicon nanowires and explored in SERS applications.48 Many of the results presented in this topic are also fundamental to developing tools for surface-enhanced spectroscopy. In this sense, the following sections will cover the use of DNA origami for single-molecule studies.
3. Single-molecule platforms
The last two sections showed how light is harvested and modified through specific and well-defined interactions either between chromophores alone, between plasmonic nanoparticles and chromophores, or between plasmonic nanoparticles alone. Even though some cases study the interactions between single molecules, many measurements were obtained under ensemble conditions, measuring an averaged property change due to the specimen's placement on the DNA origami nanostructure. The present section focuses on isolated molecules responding to an external stimulus, emphasizing radiation in its various forms.3.1. Surface-enhanced Raman scattering (SERS)
Raman spectroscopy is a characterization technique that detects the inelastic scattering of light by molecules and is commonly used to study molecular and structural vibrational fingerprints. However, Raman scattering is very weak, i.e., only 1 out of 106 scattered photons is scattered inelastically. A way to circumvent this inherent weakness is to use metallic substrates as a Raman platform. Surface-enhanced Raman scattering (SERS) is mainly based on the enhanced electromagnetic field close to the metal surface upon light excitation of the surface plasmon resonance (SPR) (Fig. 4).49,50 Such a process increases the Raman signal of molecules by many orders of magnitude, between 105 to 1011, depending on the substrate and molecule. Martin Fleischmann first observed the SERS effect in 1974,51 and later confirmed by Richard Van Duyne in 1977 from the study of adsorbed pyridine on a silver electrode surface.52 The sensitivity of SERS signals depends on the size and shape (geometry) of the plasmonic nanostructures, which, however, can be maximized to a greater extent via plasmonic coupling of nanostructures as already described above.53Due to strong signal generation, SERS was envisioned to detect single molecules and to study their behavior and dynamics. In this regard, the 2-D and 3-D DNA origami techniques offer a good platform for constructing plasmonic nanoantennas for effective and reproducible SERS.4,54 The inherent addressability and rigidity offered by DNA origami allow programmed self-assembly of different plasmonic nanostructures and the precise placement of target molecules in the region of interest.7
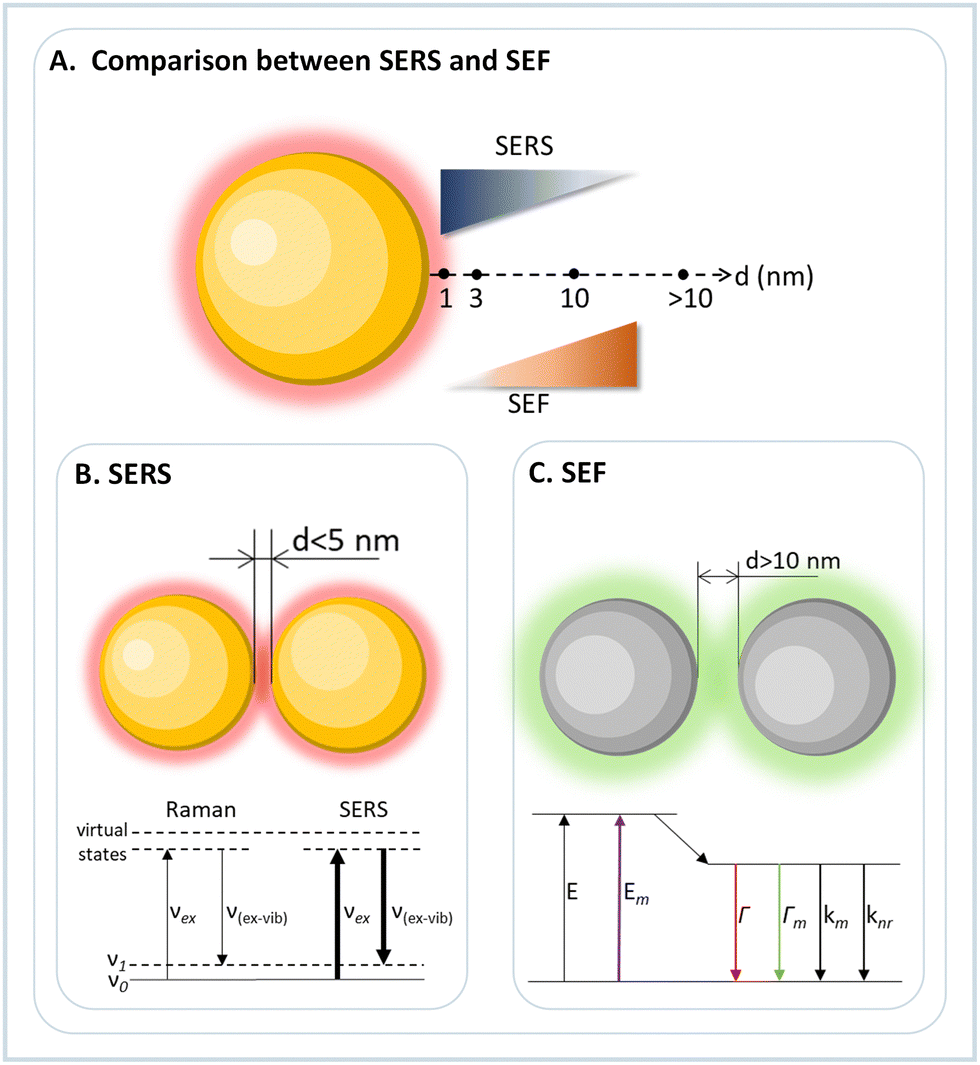 | ||
| Fig. 4 Comparison between SERS and SEF. (A) SERS and SEF intensity as a function of the distance between the probe molecule and the nanoparticle surface. At very small distances (<5 nm) SERS is dominant, as the distance increases (∼10 nm) the SEF dominates, and at very long distances (>10 nm) both effects are very weak. (B) Nanoparticle dimer with a distance between nanoparticle surface below 5 nm for SERS applications, and the energy diagram comparing the energetics of conventional Raman and SERS. (C) Nanoparticle dimer with gap distance of more than 10 nm for SEF applications, and the energy diagram showing that SEF creates new energy decay mechanisms. | ||
A very first approach towards utilizing DNA origami to construct plasmonic nanoantennas for analyzing SERS from a few molecules was demonstrated by our group in 2013.54 In this study, 40 nm spherical AuNPs coated with TAMRA modified DNA sequences were assembled using a triangular DNA origami nanostructure to obtain AuNP dimers. This ensured that approximately 40 TAMRA dyes were trapped at the hot spot contributing to the SERS signal. On the other hand, for single TAMRA dye measurements, a single dye is incorporated into a specific location of the triangular DNA origami, followed by the hybridization of 25 nm AuNPs, such that the dye is positioned between the particles, spatially close to the plasmonic hot spot. The correlated AFM imaging and Raman measurements of the same allowed assignment of SERS signals arising from a low number of individual DNA-AuNP hybrid systems, Fig. 5A.54 Those results from our group were the first ones showing the use of DNA origami for single- or few-molecule SERS. Around the same time, two papers showed DNA origami 3D nanostructures for SERS applications; both used two 40 nm AuNPs.55,56 The first used the assembly to detect DNA sequence variations by SERS, although not in the single-molecule scale. The second used the nanocavity formed between two nanoparticles to detect intercalated SYBR Gold dyes. Due to the unspecific dye incorporation method, it is estimated that around 25 molecules are possibly located in the nanocavity.56
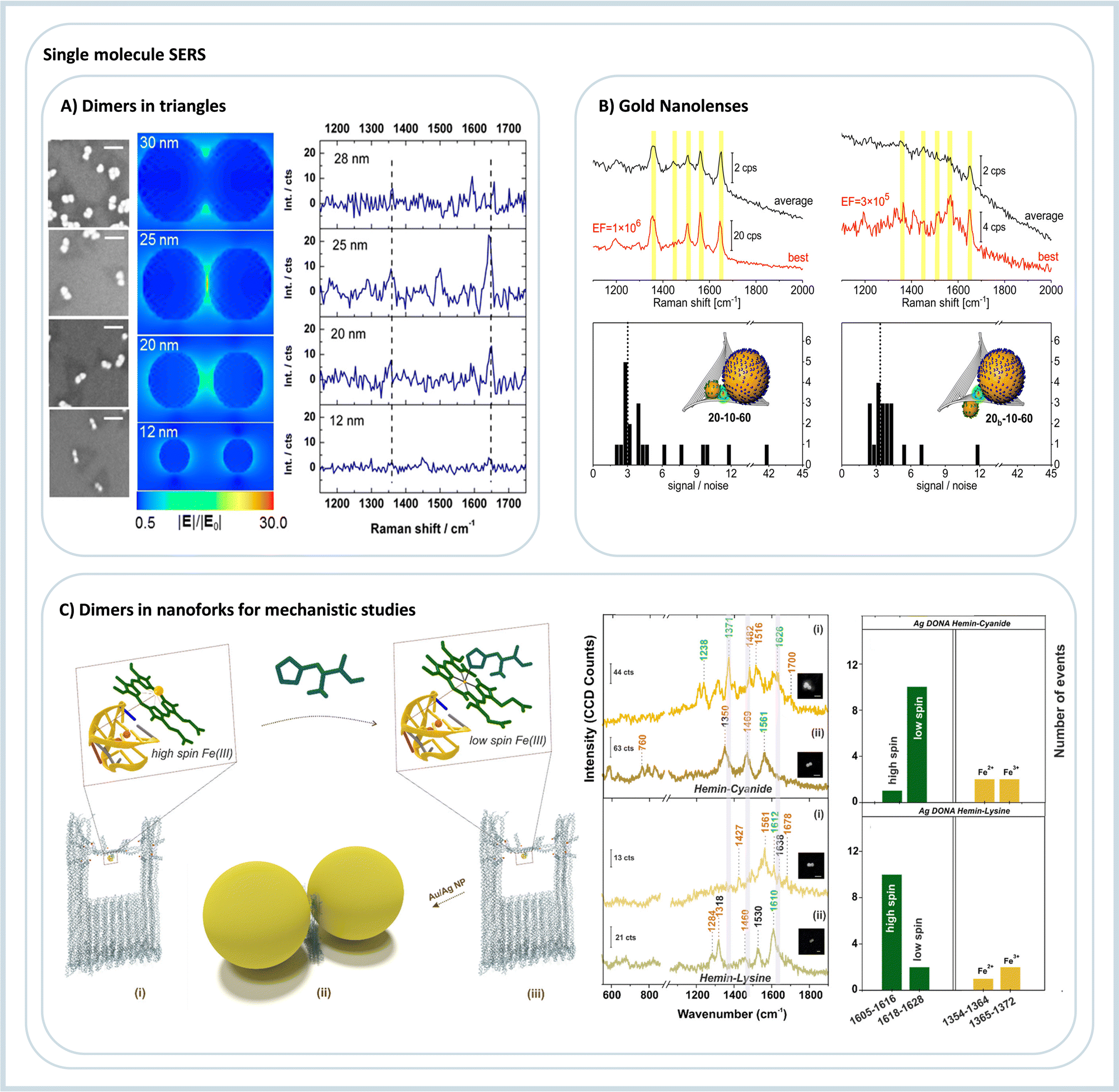 | ||
| Fig. 5 DNA origami assemblies for single-molecule SERS. (A) Nanoparticle dimers with different diameters for three-molecule SERS detection, SEM on the left, electric field simulation, and TAMRA SERS spectra. Reproduced with permission from ref. 54. Copyright 2023 American Chemical Society. (B) Two different gold nanolenses and the respective SERS spectra show enhancements in the order of 106. Reproduced with permission from ref. 57. Copyright 2023 American Chemical Society. (C) DNA origami nanoantenna using a nanofork structure to assemble nanoparticle dimers; in the example, it is used to study hemin. On the right, the single molecule SERS spectra and the statistics of event observation are due to the addition of strong and weak field ligands to the hemin. Reproduced from ref. 58 with permission from the Royal Society of Chemistry. | ||
After the initial results showed that DNA origami could generate SERS-active assemblies, optimizing such devices was the next step by first increasing the obtained signal and later stabilizing the signal over a long time. Using fully dye-covered nanoparticles arranged on DNA origami nanotriangles, we studied how the enhancement factor is distributed depending on the size of AuNPs, the chosen dye, and the excitation wavelength.59 The results showed that AuNPs with a diameter of 60 nm is optimal for obtaining SERS spectra of single dimers. Increasing the diameter to 80 nm only increased the signal slightly, but at the same time, 80 nm AuNPs are significantly more challenging to modify and stabilize. Also, we observed that 633 nm is the optimal laser wavelength for Raman excitation of Au dimers.59 We also showed graphene as a protection layer covering the AuNP dimers assembled over nanotriangles. The SERS signals of the TAMRA dyes were still observed after 800 s of continuous irradiation, showing that graphene reduced the photobleaching effect, possibly due to protection from the atmospheric oxygen and by enabling efficient heat dissipation.60 The SERS signal intensity was also optimized by electroless deposition of Ag around AuNPs.61 This procedure decreases the gap distance and also shifts the plasmonic resonance of the nanoparticles to the blue, enabling a higher signal enhancement at 532 nm, reaching enhancement factors in the order of 1010, and being able to detect single molecules efficiently.61
Assemblies composed of spherical gold and silver nanoparticles were also obtained for single-molecule SERS. For example, Tanwar and collaborators arranged gold and silver nanostars in DNA origami rectangles.62,63 Due to the spiked structure of the nanostars, the electromagnetic field is more strongly confined, and due to the DNA origami nanostructure, it is possible to space the particles in such a way that the particle-to-particle interaction is maximized. The authors showed that about 5 nm of gap distance provided a maximum enhancement factor in 1010. Such assembly was used for the single-molecule SERS analysis of dyes and single thrombin proteins.62,63 The same group used Au@Ag nanostars assembled in dimers using DNA origami to quantify pyocyanin, an important bacterial biomarker, using SERS.62–64 The detection limit was 335 pM in solution, which implies that the dimer probes only a few molecules.
The DNA origami triangle was also used to assemble silver nanocubes in six different conformations by specific patterns of DNA capture strands on the DNA origami nanotriangle.65 The authors used SERS to study the signal from fully covered particles and detect single-molecule SERS spectra of a commonly used dye. The enhancement factor of the plasmonic device was calculated to be around 107. In this work, up to four Ag nanocubes have been combined on one DNA origami, and the capabilities of DNA allow for the construction of even more complex arrangements.
Our group created plasmonic nanolenses composed of three nanoparticles of different sizes. Nanolenses, as introduced by Stockman in 2003, provides a very efficient cascaded field enhancement.66 However, the experimental methods to obtain such nanolenses are very limited, but DNA origami provides a possible route to self-assemble them in solution in a few steps.57,67 We constructed and optimized Au nanolenses for SERS analysis of dye-coated nanoparticles. By selective labeling of the outer nanoparticles, we could, for the first time, verify experimentally that the stronger hot spot is located between the medium-sized and the small particle, in line with computational predictions but in contrast to what is intuitively expected, Fig. 5B.57 With AgNPs, it was possible to achieve even stronger enhancements of more than 108, while with Au nanolenses, 106 was observed. The DNA origami technique allowed for the targeted placement of single streptavidin proteins into the hot spot with the highest signal enhancement.67 Single-molecule SERS detection of streptavidin was possible by introducing an alkyne label that allowed for an unambiguous assignment of SERS signal to single streptavidin molecules.67 Ensembles composed of four nanoparticles were obtained by Fang et al.68 The obtained metamaterials were tested to quantify the intensity of single-molecule SERS. For this, a Raman dye was placed in the region with the maximum calculated electromagnetic field enhancement, and one to twelve dyes were placed in this region. The obtained results showed that the hot spot created could fit a maximum of six dyes in a row, and with more dyes, the SERS intensity was not changing anymore. The authors also showed that the intensity of the studied spectra increased linearly between one to six dyes added in the assembly. Those results open up the possibility of quantifying molecules in the single to the few molecules regime, which poses a severe challenge because of the intrinsic heterogeneity of SERS hot spots.69
Large micrometer scale linear and two-dimensional assemblies were constructed and tested for SERS applications by Ren et al.70 The self-assembly of the metamaterial was optimized and showed a very satisfactory SERS enhancement when composed of 50 and 80 nm AuNPs. Single-molecule SERS detection was not the focus of the study. Still, the work demonstrates that DNA origami can escalate the complexity of the obtained assemblies with a feasible self-assembly process. However, the devices are still not fully operational because the hot spot is confined and not easily accessible for free molecules.
The first effort to create a clear hot spot was made by Zhan et al.,71 where gold nanotriangles were self-assembled in a bowtie format. Such assembly achieved a gap distance between nanotriangle tips of about 5 nm, with one single Cy5 dye placed in the hot spot yielding very reproducible single-molecule SERS signals. The versatility of the open hot spot was tested using two more molecules, Cy3 and an alkyne-modified DNA strand. To obtain a versatile DNA nanodevice with a cleared hot spot, we recently showed the creation of a DNA origami nanofork capable of creating nanoparticle dimers.72 In between the particles, a bridge DNA strand can be freely modified to attach different functional groups and molecules. This assembly detected single-molecule SERS spectra of three dyes and two proteins, both with Au and AgNPs. The gap distance between particles can also be modified by increasing or decreasing the length of DNA coating strands on the nanoparticles. The same DNA origami nanofork was used to detect and investigate the molecular states and the spin crossover of hemin by single-molecule SERS, Fig. 5C.73 After initial characterization of the hemin binding aptamer, its folding into a G quadruplex and the binding and spin states of the hemin, strong-field ligands that chemically interact with hemin was added to the self-assembly mixture. The obtained single-molecule SERS spectra showed the modification of the spin and oxidation state of the iron ion in the hemin complex, Fig. 5C.
DNA origami represents a very fruitful technique for fundamental studies in plasmonics because it is possible to obtain many self-similar devices with precisely arranged nanoparticles and molecular entities exploiting a simple design and a facile lab preparation. More than this, DNA offers many possibilities by using DNA-responsive properties towards external stimuli, as was tested by SERS on the single-molecule level.74–76 An important field for DNA origami plasmonic devices is the fundamental study of chemical and plasmon-induced reactions by SERS. The decomposition of DNA nucleobases by plasmonically generated hot electrons was already studied extensively. It can be expected that the same could be done at the single-molecule scale using DNA origami assemblies.77,78 In the next section, we will expand the discussion of studies from SERS to the use of surface-enhanced fluorescence.
3.2. (Surface-enhanced) fluorescence studies
The plasmonic evanescent field can not only be used to increase the light scattering from a molecule, such as observed in SERS studies, but it can also enhance fluorescence emission. However, while SERS requires a close distance between molecule and nanoparticle surface, in surface-enhanced fluorescence (SEF) the distance between nanoparticle and emitter must be larger, i.e. around 10 nm (Fig. 4). This distance is needed because the close proximity of the fluorophore with the metallic surface of the nanoparticle quenches the radiative decay. In contrast, at a larger distance, the E-field exerted by the plasmonic nanoparticle increases molecular absorption due to light confinement and opens new decay pathways. In this way, the DNA origami orthogonal positioning system allows the construction and simple optimization of devices to maximize fluorescence emission, allowing its use in single-molecule studies.The first demonstration of a plasmonic nanoantenna for fluorescence enhancement was reported by Acuña et al. in 2012.79 The authors constructed a pillar made by DNA origami with docking positions for two AuNPs and for one fluorophore, and by confocal fluorescence microscopy a fluorescence enhancement of 117-fold compared to the free fluorophore emission was observed. Like the development of SERS assemblies, the constructs for SEF evolved in obtaining the highest possible fluorescence signal and applying it to (bio)physical and (bio)analytical studies. In 2015, Puchkova et al. used the same nanopillar to assemble a dimer of 100 nm AuNPs with a gap distance of about 12 nm.80 The authors showed the single molecule detection in solution even with a high concentration of the fluorophore present. Due to the plasmonic assembly, an enhancement of 5000 was observed.
Even though AuNPs are the most common and easiest to modify, AgNPs and Au nanorods were also used in SEF studies. Ochmann et al. used one 80 nm AgNP assembled in DNA origami nanopillar to specifically detect Zika-specific DNA and RNA, with important discrimination towards non-specific oligo sequences.81 The enhancement performance of dimers of 80 nm diameter AgNP was compared to AuNP dimers of the same size by Vietz et al.82 The results showed superior performance of AgNP dimers principally in the visible range of the spectra, a region where AuNPs showed a minor enhancement. More than this, it showed comparable performance in the red part of the spectra (650 nm), enabling the same antenna to be used with broadband excitation. Using DNA nanorectangles to assemble Au nanorods, Zhang et al. showed enhancements in the order of 470 fold with a gap distance of about 6.1 nm between nanorods tips.83 Also, using Au nanorods assembled using a DNA origami nanopillar, Trofymchuk et al. showed an enhanced fluorescence emission in the near IR (760 nm) by three orders of magnitude compared to an assembly using 40 nm spherical AuNPs.84 Those efforts increase the possibilities of controllably using SEF as an analytical tool; however, in many of those works, the hot spot is not accessible to diffusing molecules.
To overcome the limitation of hot-spot accessibility, Trofymchuk and Glembockyte et al. developed a new DNA origami nanotower where the recognition element is placed between the nanoparticles keeping an open space in which molecules can diffuse into.85 The authors showed the use of such assembly to detect labeled single molecule DNA, in and out of the hot-spot, and as a proof-of-principle, demonstrated the use of the construct to detect a single gene sequence qualitatively. Further optimization led to the development of a DNA nanotrident. In this case, an even larger open diffusion space is obtained without increasing the nanoparticles’ distance, resulting in a design with enhancements in the order of 76 fold.86 At the same time, the larger area was used to detect DNA single strands with a length of 151 nucleotides. Such open space can further contribute to studies on biophysics since different active biomolecules, such as proteins, can be placed there to study dynamic processes. Fig. 6 shows the DNA origami design for the three generations of SEF nanoantennas constructed as the nanopillar, the nanotower, and the nanotrident.
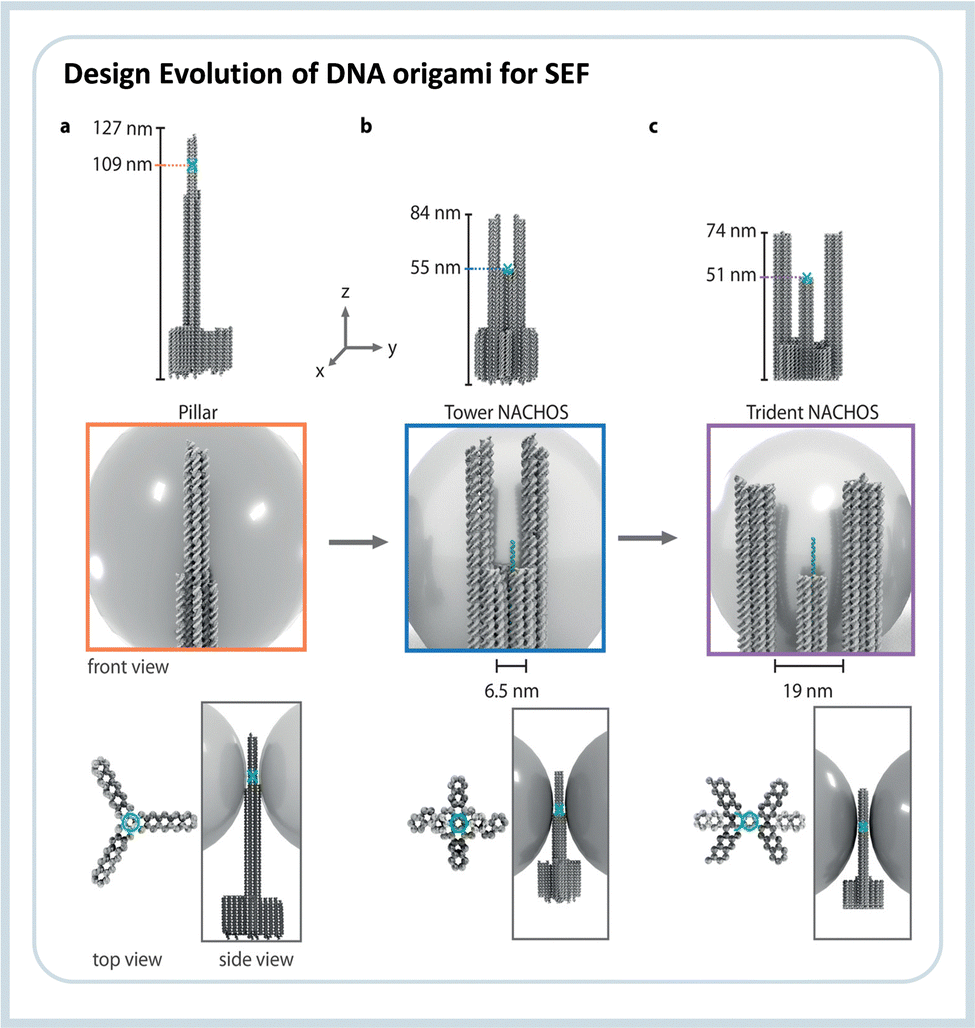 | ||
| Fig. 6 Evolution line of the DNA origami design used in SEF studies. The first structure (i) was called “nanopillar” which did not allow the entrance of analytes, the second (ii) called “nanotower” presented an open region of about 6.5 nm, and the third and newest design (iii) the “nanotrident” has an opening of about 19 nm, which is enough to accommodate proteins. Reproduced from ref. 86 with permission from Wiley. | ||
One example of a biophysical study using SEF enabled by DNA origami assemblies was reported by Kaminska et al. in 2018.87 Using a DNA origami nanopillar, the natural light-harvesting complex of peridinin–chlorophyll α-protein was placed in the hot-spot of a dimer structure composed of both Au and AgNP. Even though there is no new biophysical result, the assembly showed that DNA origami could be used to spectroscopically study biomolecules in their native state, principally to develop bioinspired plasmonic light harvesting constructs further. The analytical use of the DNA origami nanotower was also demonstrated by the single antibody detection in the nanocavity. The mechanism of detection involved the opening of a DNA nanoswitch, and after an incubation time of 20 min it was possible to detect the signal from concentrations as low as 1.4 nM.88 Those results open up many possibilities for studying different light-induced processes in the nanocavity of the DNA origami construct, and in the future many more single-molecule mechanistic studies are expected to be realized using such nanostructures.
DNA origami nanostructures are already very well developed, and most of their properties are fundamentally studied. In the future, we expect that more studies involving dynamic processes and mechanisms will be solved using single-molecule SERS and SEF. So far, we showed the application of DNA origami to organize different molecules and nanoparticles to study processes by optical spectroscopy, which is only possible due to the use of plasmonic nanoparticles. In the next section, the use of DNA origami to study DNA radiation damage by AFM will be explained.
3.3. Atomic force microscopy: quantifying DNA radiation damage
DNA origami platforms can be easily analyzed by AFM, which is, on the one hand, routinely used to characterize the integrity of single DNA origami nanostructures.89,90 On the other hand, the lateral resolution of AFM is sufficient to resolve the positions of molecular modifications on the DNA origami structures, most important proteins, and bulky DNA structures. Consequently, AFM is ideally suited to identify the presence of such entities at specific positions on the DNA origami platforms. This can be exploited to study, e.g., chemical and biochemical processes at a single-molecule level,91,92 which has been reviewed in detail before.9AFM is a type of scanning probe microscopy that relies on the sensitive detection of forces between the tip and the sample surface. AFM tapping mode (AFM-TM) works by oscillating the cantilever near its resonant frequency close to the surface of the sample and lightly tapping the surface of the sample during each oscillation. The amplitude of the cantilever is monitored, and this information is processed to create a topographical image of the sample surface. AFM-TM is thus less likely to damage soft samples compared to other commonly used AFM imaging modes, e.g., contact mode. Here, we focus on one example of Lab-on-a-DNA origami, where the interaction of DNA origami with different kinds of radiation sources is studied. The basic experimental scheme used for these studies is illustrated in Fig. 7A.93 Triangular DNA origami structures are mainly used because they are the stiffest 2D DNA origami structure that does not tend to form aggregates.94 The target DNA structures are extended from staple strands of the DNA origami and are labeled with biotin. After deposition on a solid substrate, the samples are irradiated, and after rinsing, the structures are incubated with streptavidin to visualize the remaining intact DNA structures in the AFM images. Radiation-induced strand breaks in the target sequences result in missing streptavidin labels. The method allows for absolute quantification of strand break yields in terms of strand break cross sections. The absolute quantification of radiation-induced DNA strand breaks with other methods is very challenging, and another advantage of the use of DNA origami platforms is the possibility of comparing two different DNA sequences in a single irradiation experiment. A complementary approach has been developed by Sala et al.95 using a DNA origami nanoframe to quantify double strand breaks in DNA that are spanned between two sides of the frame and exposed to different types of radiation, as illustrated in Fig. 8A. The double-stranded DNA is directly visualized by AFM, which also allows for direct visualization of strand breaks without further modification after the irradiation. The irradiation can be performed in solution (and subsequent deposition of the DNA nanostructures on a suitable surface) and directly on a substrate. As shown in Fig. 8B, this approach has been recently used to study the effect of high-energy electrons (16 MeV) on the double-strand break yields for specific DNA sequences. Such high-energy radiation is used in cancer therapy to reduce the tumor tissue with high local control of dose deposition.
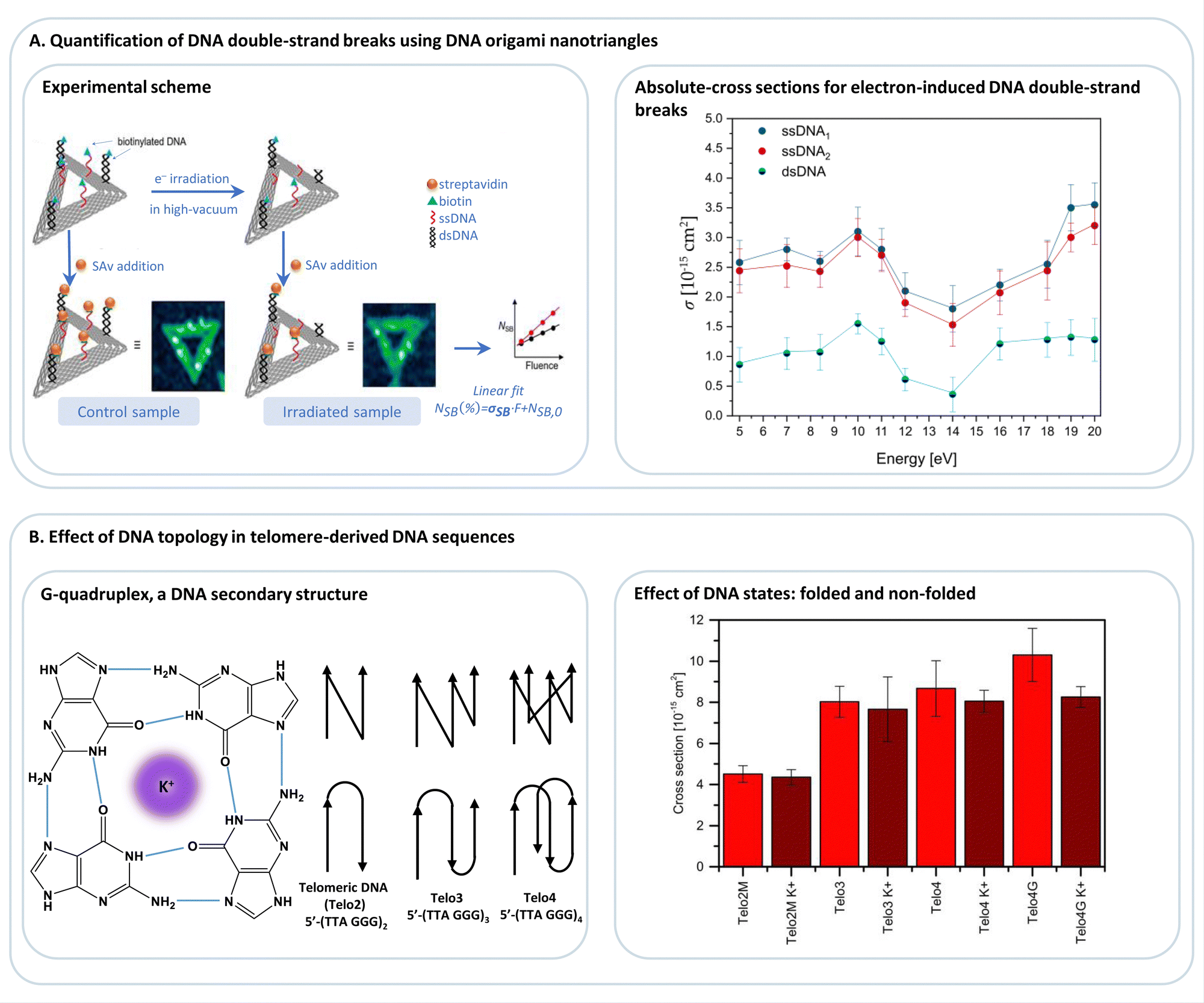 | ||
| Fig. 7 (A) DNA origami nanotriangles serve as a platform to expose biotinylated DNA sequences, such as single-stranded or double-stranded DNA (ssDNA, dsDNA; respectively), to low-energy electrons (<20 eV), and thus allowing for the determination of absolute cross-sections for electron-induced DNA strand breakage. Adapted with permission from ref. 93. (B) Studying the effect of DNA topology on electron-induced DNA single-strand breaks in telomere-derived DNA sequences (Telo2, Telo3, Telo4), which are known to fold into G-quadruplexes in the presence of monocations, e.g., K+, (Telo2MK+, Telo3K+, Telo4K+, Telo4GK+). Adapted from ref. 96 with permission from Wiley. | ||
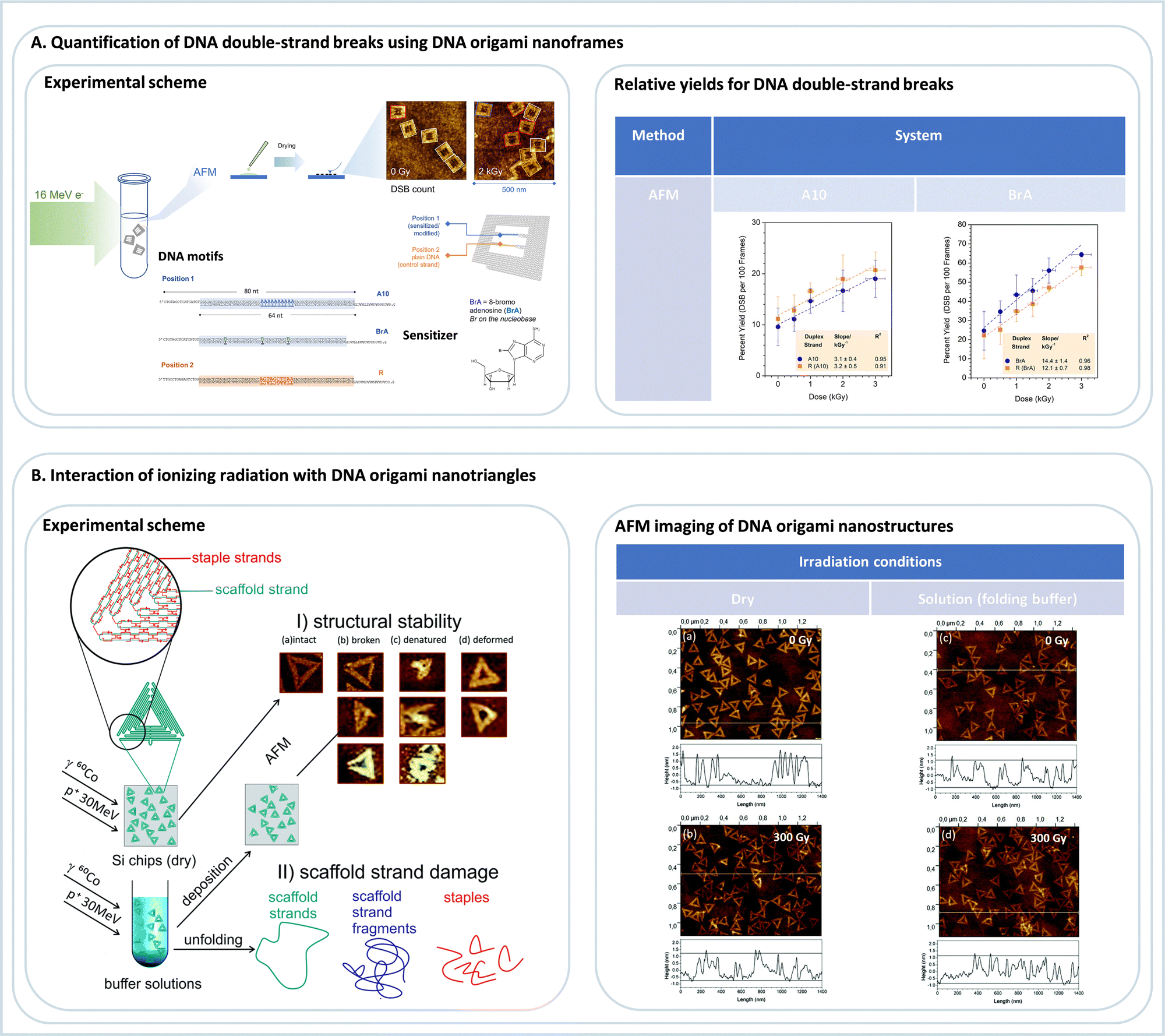 | ||
| Fig. 8 (A) Quantification of DNA double-strand breaks using DNA origami nanoframes. Following the irradiation of a buffer solution containing DNA origami nanoframes with 16 MeV electrons, irradiated samples were deposited on Si chips for subsequent AFM analysis. Relative yields for DNA double-strand breaks were thus determined for two different DNA sequences as a function of the dose: a control sequence A10 and a sensitized sequence, which incorporates 8-bromoadenine (8brA). Adapted from ref. 95. Copyright 2023 from American Chemical Society (B) studying the interaction of different ionizing radiations, such as gamma rays or fast protons with DNA origami nanotriangles in either dried or in buffer conditions. Subsequently, the stability of DNA origami nanostructures exposed to radiation was assessed by (i) AFM imaging or (ii) gel electrophoresis analysis (not shown here). Adapted from ref. 97 with permission from Royal Society of Chemistry. | ||
Most of the cellular damage induced by high-energy photons or particles is not directly due to the primary particles but due to the secondarily generated particles that are mostly products of water radiolysis. The most important secondary particles are hydroxyl radicals and low-energy electrons.98 Low-energy electrons have a kinetic energy distribution that peeks around 10 eV and reaches very low energies close to zero eV.99 As these energies are close to or even below the ionization energies of DNA, they have been regarded as irrelevant for DNA radiation damage for a very long time. However, it was shown that low-energy electrons could efficiently induce several types of DNA damage, such as base loss, single and double-strand breaks, and interstrand cross-links by dissociative electron attachment (DEA) processes.100–103 In DEA, the electron occupies a formerly empty molecular orbital of the DNA nucleobases or the DNA backbone, which can eventually weaken and cleave a C–O bond between the sugar and phosphate bond in the DNA backbone, which represents a DNA strand break.104–106
DNA origami platforms have been used to study the low-energy electron-induced strand breakage for a broad range of DNA sequences and electron energies. The energy dependence of single and double-strand breaks shows the characteristic resonant features of the DEA process (Fig. 7A) that are peaking at 10 eV for DNA. The experiments using DNA origami platforms have, in general, showing that there is not a pronounced dependence of low-energy electron-induced strand breakage on the DNA sequence. The only exception revealed so far is that the G-rich telomeric repeat unit 5′-d(TTAGGG) is more prone to electron-induced strand breakage than a sequence with the same number of each nucleobase but randomly mixed.96 This was explained by a polarization of the G stack by the neighboring A, leading to stronger electron attraction. Telomeric DNA is found at the ends of chromosomes, and their length determines the life span of a cell. Four repeat units can fold into a G quadruplex structure (Fig. 7B) in the presence of K+ ions and other mono- and divalent ions.28,107 The G quadruplex stabilizes the telomeric DNA towards low-energy electrons. Nevertheless, it was suggested to use G-quadruplex targeting molecules to exploit the intrinsic sensitivity of the telomeric G-rich sequence towards radiation to treat cancer cells.
Conventional cancer radiation therapy is often combined with the administration of radiosensitizing therapeutics,108,109 such as fluorinated nucleosides to show a supra-additive effect, i.e., the combination of radiation administration and chemotherapy shows a synergy and better treatment results than each treatment alone.110 In the case of halogenated nucleosides, the radiosensitizing effect can at least partly be ascribed to a higher reactivity towards secondary electrons compared to non-halogenated nucleosides. This effect can be demonstrated accurately using the DNA origami technique because two different, but well-defined target sequences can be compared in a single irradiation experiment, e.g., one sequence modified with a radiosensitizer and another without this modification. After irradiation at a specific electron energy, the yield of strand breaks can be determined in terms of absolute strand breaks for both sequences, and the ratio of the strand break cross sections of modified and non-modified sequences corresponds to the enhancement factor that is characteristic for the respective radiosensitizer. Using the DNA origami technique, enhancement factors for 5-fluorouracil,111,112 2-fluoroadenine,113 and 8-bromoadenine114 containing sequences have been determined at various electron energy, yielding values between 1.5 and 2.0. This demonstrated that the DNA origami technique could be used to accurately assess the effectivity of a radiosensitizing compound to identify potential new therapeutics but also to understand the radiosensitizing mechanism in more detail.
By using the DNA nanoframes an interesting proximity effect was observed with 8-bromoadenine. Irradiation with 16 MeV electrons does not only lead to enhanced strand breakage in the modified DNA double strand but also in a larger number of strand breaks in the neighboring, unmodified control strand.95 This is probably due to a generation of secondary reactive products from 8-bromoadenine that can also damage the surrounding. Such effects can be very well studied using DNA origami platforms and are important for developing efficient radiosensitizers for radiation therapy and need further fundamental investigations in the future.
In a series of studies,115–117 the DNA origami technique was also used to study the interactions of vacuum-UV (VUV) photons with well-defined DNA sequences attached to DNA origami. VUV photons are interesting because they possess energy around the ionization threshold of DNA and help to gain a fundamental understanding of photon-induced DNA damage processes. It was found that strand breaks can be generated even by photons having energy as low as 6.5 eV, and the damage yield increases steeply above the ionization energy of the nucleobases, which is at 8.4 eV for A. The advantage of using the DNA origami technique in this context is the absolute quantification of strand break cross sections. These values can be compared with the absolute cross sections for absorption, and the ratio of both values yields the quantum yield for strand breakage. It was found that the quantum yield for strand breakage induced by 8.4 eV photons is as high as 50%. In further studies, the dependence of photon-induced strand breakage on the DNA sequence and, in particular, the effect of the photosensitizer 5-bromouracil was studied using DNA triangles.
Very recently, the radiation stability of the DNA origami triangles themselves was studied using 60Co γ-rays (1.17 and 1.33 MeV) and 30 MeV proton beams (Fig. 8A).97 It was found that the DNA origami triangles exhibit significantly higher radiation stability than the double-stranded M13mp18 DNA. Even under radiation doses as high as 300 Gy, as shown in Fig. 8B, the DNA origami structures remained largely intact, which was ascribed to the strongly interconnected and dense DNA structure.
4. Conclusion and future directions
The Lab-on-a-DNA origami approach combines state-of-the-art nanoengineering tools with highly sensitive detection techniques. It provides, in this way, novel possibilities for the interrogation of nanoscale systems and for creating new concepts for sensing and energy conversion.In the future, the single-molecule detection methods by SERS or SEF need to be developed into a routine tool for biological and chemical sensing, but also for fundamental biophysical studies. An interesting possibility for sensing is the interfacing of DNA origami platforms with other technologies, such as optical fibers. For such approaches, the surface anchoring of DNA origami and their stability must be improved. For the latter, several strategies have been suggested, e.g., UV cross-linking, enzymatic ligation, and polymer coating.118–121
Apart from sensing, the Lab-on-a-DNA origami approach allows for unprecedented fundamental biological, physical and chemical studies, and especially SERS provides the possibility to obtain direct chemical information of an analyte to study molecular properties and chemical reaction pathways as it was recently demonstrated using hemin.58 Despite the potential of SERS and SEF for biophysical studies, fundamental interactions of single molecules and plasmonic nanostructures need to be understood in more detail. Other highly interesting fields of application for the Lab-on-a-DNA origami approach are e.g. enzymatic reaction cascades122 or the interrogation of biomolecular systems by force measurements.123
The Lab-on-a-DNA origami approach already allowed us to collect detailed information about the sequence-dependence of DNA strand breaks. Still, this field is just at the beginning of what is possible in the future. New and optimized DNA origami structures can be developed and applied to study the effect of newly designed therapeutically relevant radiosensitizers using a broader range of primary radiations. Moreover, the investigation of radiation damage mechanisms to other therapeutically relevant approaches, such as photodynamic and photothermal therapy (PDT and PTT, respectively), can be extended. Very recently, the DNA single and double strand breaks induced by irradiation of a molecular ruby have been quantified, which opens a new field of application for the Lab-on-a-DNA origami approach.124
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support by the European Research Council (ERC; consolidator grant no. 772752).References
- N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2017, 3, 17068 CrossRef.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- F. Hong, F. Zhang, Y. Liu and H. Yan, Chem. Rev., 2017, 117, 12584–12640 CrossRef CAS PubMed.
- K. Tapio and I. Bald, Multifunct. Mater., 2020, 3, 032001 CrossRef CAS.
- Q. Jiang, S. Liu, J. Liu, Z. Wang and B. Ding, Adv. Mater., 2019, 31, 1804785 CrossRef CAS PubMed.
- S. Kogikoski, W. J. Paschoalino and L. T. Kubota, TrAC, Trends Anal. Chem., 2018, 108, 88–97 CrossRef CAS.
- A. Kuzyk, R. Jungmann, G. P. Acuna and N. Liu, ACS Photonics, 2018, 5, 1151–1163 CrossRef CAS PubMed.
- Y. Ke, S. Lindsay, Y. Chang, Y. Liu and H. Yan, Science, 2008, 319, 180–183 CrossRef CAS PubMed.
- I. Bald and A. Keller, Molecules, 2014, 19, 13803–13823 CrossRef PubMed.
- V. Glembockyte, L. Grabenhorst, K. Trofymchuk and P. Tinnefeld, Acc. Chem. Res., 2021, 54, 3338–3348 CrossRef CAS PubMed.
- M. R. Jones, N. C. Seeman and C. A. Mirkin, Science, 2015, 347, 1260901 CrossRef PubMed.
- S. M. Douglas, H. Dietz, T. Liedl, B. Högberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS PubMed.
- H. Dietz, S. M. Douglas and W. M. Shih, Science, 2009, 325, 725–730 CrossRef CAS PubMed.
- L. L. Ong, N. Hanikel, O. K. Yaghi, C. Grun, M. T. Strauss, P. Bron, J. Lai-Kee-Him, F. Schueder, B. Wang, P. Wang, J. Y. Kishi, C. Myhrvold, A. Zhu, R. Jungmann, G. Bellot, Y. Ke and P. Yin, Nature, 2017, 552, 72–77 CrossRef CAS PubMed.
- K. F. Wagenbauer, C. Sigl and H. Dietz, Nature, 2017, 552, 78–83 CrossRef CAS PubMed.
- F. Praetorius, B. Kick, K. L. Behler, M. N. Honemann, D. Weuster-Botz, H. Dietz and P. E. Coli, Nature, 2017, 552, 84–87 CrossRef CAS PubMed.
- C. M. Wintersinger, D. Minev, A. Ershova, H. M. Sasaki, G. Gowri, J. F. Berengut, F. E. Corea-Dilbert, P. Yin and W. M. Shih, Nat. Nanotechnol., 2022, 2022, 1–9 Search PubMed.
- G. Tikhomirov, P. Petersen and L. Qian, Nature, 2017, 552, 67–71 CrossRef CAS PubMed.
- S. Dey, C. Fan, K. v Gothelf, J. Li, C. Lin, L. Liu, N. Liu, M. A. D. Nijenhuis, B. Saccà, F. C. Simmel, H. Yan and P. Zhan, Nat. Rev. Methods Primers, 2021, 1, 1–24 CrossRef.
- M. Glaser, S. Deb, F. Seier, A. Agrawal, T. Liedl, S. Douglas, M. K. Gupta and D. M. Smith, Molecules, 2021, 26, 2287 CrossRef CAS PubMed.
- L. Olejko, P. J. Cywiński and I. Bald, Nanoscale, 2016, 8, 10339–10347 RSC.
- X. Zhou, D. Satyabola, H. Liu, S. Jiang, X. Qi, L. Yu, S. Lin, Y. Liu, N. W. Woodbury and H. Yan, Angew. Chem., Int. Ed., 2022, 61, e202211200 CAS.
- M. Madsen, M. R. Bakke, D. A. Gudnason, A. F. Sandahl, R. A. Hansen, J. B. Knudsen, A. L. B. Kodal, V. Birkedal and K. v Gothelf, ACS Nano, 2021, 15, 9404–9411 CrossRef CAS PubMed.
- P. K. Dutta, R. Varghese, J. Nangreave, S. Lin, H. Yan and Y. Liu, J. Am. Chem. Soc., 2011, 133, 11985–11993 CrossRef CAS PubMed.
- I. H. Stein, V. Schüller, P. Böhm, P. Tinnefeld and T. Liedl, Chem. Phys. Chem., 2011, 12, 689–695 CrossRef CAS PubMed.
- I. H. Stein, C. Steinhauer and P. Tinnefeld, J. Am. Chem. Soc., 2011, 133, 4193–4195 CrossRef CAS PubMed.
- L. Olejko, P. J. Cywinski and I. Bald, Angew. Chem., Int. Ed., 2015, 54, 673–677 CAS.
- L. Olejko, A. Dutta, K. Shahsavar and I. Bald, Int. J. Mol. Sci., 2022, 23, 12206 CrossRef CAS PubMed.
- Y. Choi, L. Kotthoff, L. Olejko, U. Resch-Genger and I. Bald, ACS Appl. Mater. Interfaces, 2018, 10, 23295–23302 CrossRef CAS PubMed.
- E. A. Hemmig, C. Creatore, B. Wünsch, L. Hecker, P. Mair, M. A. Parker, S. Emmott, P. Tinnefeld, U. F. Keyser and A. W. Chin, Nano Lett., 2016, 16, 2369–2374 CrossRef CAS PubMed.
- L. Olejko and I. Bald, RSC Adv., 2017, 7, 23924–23934 RSC.
- F. Nicoli, A. Barth, W. Bae, F. Neukirchinger, A. H. Crevenna, D. C. Lamb and T. Liedl, ACS Nano, 2017, 11, 11264–11272 CrossRef CAS PubMed.
- X. Zhou, H. Liu, F. Djutanta, D. Satyabola, S. Jiang, X. Qi, L. Yu, S. Lin, R. F. Hariadi, Y. Liu, N. W. Woodbury and H. Yan, Chem, 2022, 8, 2442–2459 CAS.
- X. Zhou, S. Mandal, S. Jiang, S. Lin, J. Yang, Y. Liu, D. G. Whitten, N. W. Woodbury and H. Yan, J. Am. Chem. Soc., 2019, 141, 8473–8481 CrossRef CAS PubMed.
- X. Wang, C. Li, D. Niu, R. Sha, N. C. Seeman and J. W. Canary, Nano Lett., 2018, 18, 2112–2115 CrossRef CAS PubMed.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- D. Devasia, A. Das, V. Mohan and P. K. Jain, Annu. Rev. Phys. Chem., 2021, 72, 423–443 CrossRef CAS PubMed.
- G. P. Acuna, M. Bucher, I. H. Stein, C. Steinhauer, A. Kuzyk, P. Holzmeister, R. Schreiber, A. Moroz, F. D. Stefani, T. Liedl, F. C. Simmel and P. Tinnefeld, ACS Nano, 2012, 6, 3189–3195 CrossRef CAS PubMed.
- A. Samanta, Y. Zhou, S. Zou, H. Yan and Y. Liu, Nano Lett., 2014, 14, 5052–5057 CrossRef CAS PubMed.
- M. Raab, I. Jusuk, J. Molle, E. Buhr, B. Bodermann, D. Bergmann, H. Bosse and P. Tinnefeld, Sci. Rep., 2018, 8, 1–11 CAS.
- M. Scheckenbach, J. Bauer, J. Zähringer, F. Selbach and P. Tinnefeld, APL Mater., 2020, 8, 110902 CrossRef CAS.
- N. T. Anderson, S. Ren, J. Chao, P. H. Dinolfo and X. Wang, ACS Appl. Nano Mater., 2019, 2, 5563–5572 CrossRef CAS.
- N. Aissaoui, K. Moth-Poulsen, M. Käll, P. Johansson, L. M. Wilhelmsson and B. Albinsson, Nanoscale, 2017, 9, 673–683 RSC.
- K. Martens, F. Binkowski, L. Nguyen, L. Hu, A. O. Govorov, S. Burger and T. Liedl, Nat. Commun., 2021, 12, 1–6 CrossRef PubMed.
- K. Vogele, J. List, G. Pardatscher, N. B. Holland, F. C. Simmel and T. Pirzer, ACS Nano, 2016, 10, 11377–11384 CrossRef CAS PubMed.
- F. N. Gür, C. P. T. McPolin, S. Raza, M. Mayer, D. J. Roth, A. M. Steiner, M. Löffler, A. Fery, M. L. Brongersma, A. v Zayats, T. A. F. König and T. L. Schmidt, Nano Lett., 2018, 18, 7323–7329 CrossRef PubMed.
- E. M. Roller, L. v Besteiro, C. Pupp, L. K. Khorashad, A. O. Govorov and T. Liedl, Nat. Phys., 2017, 13, 761–765 Search PubMed.
- A. Moeinian, F. N. Gür, J. Gonzalez-Torres, L. Zhou, V. D. Murugesan, A. D. Dashtestani, H. Guo, T. L. Schmidt and S. Strehle, Nano Lett., 2019, 19, 1061–1066 CrossRef CAS PubMed.
- R. Pilot, R. Signorini, C. Durante, L. Orian, M. Bhamidipati and L. Fabris, Biosensors, 2019, 9, 57 CrossRef CAS PubMed.
- R. Pilot, R. Signorini, C. Durante, L. Orian, M. Bhamidipati and L. Fabris, Biosensors, 2019, 9, 57 CrossRef CAS PubMed.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- D. L. Jeanmaire and R. P. van Duyne, J. Electroanal. Chem. Interfacial Electrochem., 1977, 84, 1–20 CrossRef CAS.
- J. Langer, D. J. de Aberasturi, J. Aizpurua, R. A. Alvarez-Puebla, B. Auguié, J. J. Baumberg, G. C. Bazan, S. E. J. Bell, A. Boisen, A. G. Brolo, J. Choo, D. Cialla-May, V. Deckert, L. Fabris, K. Faulds, F. Javier García de Abajo, R. Goodacre, D. Graham, A. J. Haes, C. L. Haynes, C. Huck, T. Itoh, M. Käll, J. Kneipp, N. A. Kotov, H. Kuang, E. C. le Ru, H. K. Lee, J. F. Li, X. Y. Ling, S. A. Maier, T. Mayerhöfer, M. Moskovits, K. Murakoshi, J. M. Nam, S. Nie, Y. Ozaki, I. Pastoriza-Santos, J. Perez-Juste, J. Popp, A. Pucci, S. Reich, B. Ren, G. C. Schatz, T. Shegai, S. Schlücker, L. L. Tay, K. George Thomas, Z. Q. Tian, R. P. van Duyne, T. Vo-Dinh, Y. Wang, K. A. Willets, C. Xu, H. Xu, Y. Xu, Y. S. Yamamoto, B. Zhao and L. M. Liz-Marzán, ACS Nano, 2020, 14, 28–117 CrossRef CAS PubMed.
- J. Prinz, B. Schreiber, L. Olejko, J. Oertel, J. Rackwitz, A. Keller and I. Bald, J. Phys. Chem. Lett., 2013, 4, 4140–4145 CrossRef CAS.
- V. v Thacker, L. O. Herrmann, D. O. Sigle, T. Zhang, T. Liedl, J. J. Baumberg and U. F. Keyser, Nat. Commun., 2014, 5, 1–7 Search PubMed.
- P. Kühler, E. M. Roller, R. Schreiber, T. Liedl, T. Lohmüller and J. Feldmann, Nano Lett., 2014, 14, 2914–2919 CrossRef PubMed.
- C. Heck, J. Prinz, A. Dathe, V. Merk, O. Stranik, W. Fritzsche, J. Kneipp and I. Bald, ACS Photonics, 2017, 4, 1123–1130 CrossRef CAS.
- A. Dutta, K. Tapio, A. Suma, A. Mostafa, Y. Kanehira, V. Carnevale, G. Bussi and I. Bald, Nanoscale, 2022, 14, 16467–16478 RSC.
- S. Kogikoski, K. Tapio, R. E. von Zander, P. Saalfrank and I. Bald, Molecules, 2021, 26, 1684 CrossRef CAS PubMed.
- J. Prinz, A. Matkovic, J. Pes, I. Bald, G. J. Nanoparticles Prinz, I. Bald, A. Matkovic, J. Pes and R. Gajic, Small, 2016, 12, 5458–5467 CrossRef CAS PubMed.
- J. Prinz, C. Heck, L. Ellerik, V. Merk and I. Bald, Nanoscale, 2016, 8, 5612–5620 RSC.
- S. Tanwar, K. K. Haldar and T. Sen, J. Am. Chem. Soc., 2017, 139, 17639–17648 CrossRef CAS PubMed.
- S. Tanwar, V. Kaur, G. Kaur and T. Sen, J. Phys. Chem. Lett., 2021, 12, 8141–8150 CrossRef CAS PubMed.
- V. Kaur, S. Tanwar, G. Kaur and T. Sen, Chem. Phys. Chem., 2021, 22, 160–167 CrossRef CAS PubMed.
- R. Niu, F. Gao, D. Wang, D. Zhu, S. Su, S. Chen, L. Yuwen, C. Fan, L. Wang and J. Chao, ACS Nano, 2022, 16, 14622–14631 CrossRef CAS PubMed.
- K. Li, M. I. Stockman and D. J. Bergman, Phys. Rev. Lett., 2003, 91, 227402 CrossRef PubMed.
- C. Heck, Y. Kanehira, J. Kneipp and I. Bald, Angew. Chem., Int. Ed., 2018, 57, 7444–7447 CrossRef CAS PubMed.
- W. Fang, S. Jia, J. Chao, L. Wang, X. Duan, H. Liu, Q. Li, X. Zuo, L. Wang, L. Wang, N. Liu and C. Fan, Sci. Adv., 2019, 5, eaau4506 CrossRef PubMed.
- G. Q. Wallace and D. Graham, Nat. Rev. Chem., 2022, 6, 842–843 CrossRef.
- S. Ren, J. Wang, C. Song, Q. Li, Y. Yang, N. Teng, S. Su, D. Zhu, W. Huang, J. Chao, L. Wang and C. Fan, Research, 2019, 2019, 1–10 CrossRef PubMed.
- P. Zhan, T. Wen, Z. Wang, Y. He, J. Shi, T. Wang, X. Liu, G. Lu and B. Ding, Angew. Chem., Int. Ed., 2018, 130, 2896–2900 CrossRef.
- K. Tapio, A. Mostafa, Y. Kanehira, A. Suma, A. Dutta and I. Bald, ACS Nano, 2021, 15, 7065–7077 CrossRef CAS PubMed.
- A. Dutta, K. Tapio, A. Suma, A. Mostafa, Y. Kanehira, V. Carnevale, G. Bussi and I. Bald, Nanoscale, 2022, 14, 16467–16478 RSC.
- B. Liu, S. Ren, Y. Xing, N. Teng, J. Wang, D. Zhu, S. Su, H. Peng, L. Ang, L. Wang, J. Chao, B. Liu, S. Ren, Y. Xing, N. Teng, J. Wang, D. Zhu, S. Su, L. Wang, P. C. Hao and H. Peng, ChemNanoMat, 2017, 3, 760–763 CrossRef CAS.
- S. Simoncelli, E. M. Roller, P. Urban, R. Schreiber, A. J. Turberfield, T. Liedl and T. Lohmüller, ACS Nano, 2016, 10, 9809–9815 CrossRef CAS PubMed.
- B. Huo, L. Xia, Z. Gao, G. Li and Y. Hu, Anal. Chem., 2022, 94, 11889–11897 CrossRef CAS PubMed.
- S. Kogikoski, A. Dutta and I. Bald, ACS Nano, 2021, 15, 20562–20573 CrossRef CAS PubMed.
- A. Dutta, R. Schurmann, S. Kogikoski, N. S. Mueller, S. Reich and I. Bald, ACS Catal., 2021, 11, 8370–8381 CrossRef CAS PubMed.
- G. P. Acuna, F. M. Möller, P. Holzmeister, S. Beater, B. Lalkens and P. Tinnefeld, Science, 2012, 338, 506–510 CrossRef CAS PubMed.
- A. Puchkova, C. Vietz, E. Pibiri, B. Wünsch, M. Sanz Paz, G. P. Acuna and P. Tinnefeld, Nano Lett., 2015, 15, 8354–8359 CrossRef CAS PubMed.
- S. E. Ochmann, C. Vietz, K. Trofymchuk, G. P. Acuna, B. Lalkens and P. Tinnefeld, Anal. Chem., 2017, 89, 13000–13007 CrossRef CAS PubMed.
- C. Vietz, I. Kaminska, M. Sanz Paz, P. Tinnefeld and G. P. Acuna, ACS Nano, 2017, 11, 4969–4975 CrossRef CAS PubMed.
- T. Zhang, N. Gao, S. Li, M. J. Lang and Q. H. Xu, J. Phys. Chem. Lett., 2015, 6, 2043–2049 CrossRef CAS PubMed.
- K. Trofymchuk, K. Kołątaj, V. Glembockyte, F. Zhu, G. P. Acuna, T. Liedl and P. Tinnefeld, ACS Nano, 2022, 17, 1327–1334 Search PubMed.
- K. Trofymchuk, V. Glembockyte, L. Grabenhorst, F. Steiner, C. Vietz, C. Close, M. Pfeiffer, L. Richter, M. L. Schütte, F. Selbach, R. Yaadav, J. Zähringer, Q. Wei, A. Ozcan, B. Lalkens, G. P. Acuna and P. Tinnefeld, Nat. Commun., 2021, 12, 1–8 CrossRef PubMed.
- C. Close, K. Trofymchuk, L. Grabenhorst, B. Lalkens, V. Glembockyte, P. Tinnefeld, C. Close, K. Trofymchuk, L. Grabenhorst, V. Glembockyte, P. Tinnefeld, B. Lalkens and L. Kamp, Adv. Mater. Interfaces, 2022, 9, 2200255 CrossRef.
- I. Kaminska, J. Bohlen, S. Mackowski, P. Tinnefeld and G. P. Acuna, ACS Nano, 2018, 12, 1650–1655 CrossRef CAS PubMed.
- M. Pfeiffer, K. Trofymchuk, S. Ranallo, F. Ricci, F. Steiner, F. Cole, V. Glembockyte and P. Tinnefeld, iScience, 2021, 9, 103072 CrossRef PubMed.
- S. Ramakrishnan, H. Ijäs, V. Linko and A. Keller, Comput. Struct. Biotechnol. J., 2018, 16, 342–349 CrossRef CAS PubMed.
- S. Ramakrishnan, G. Krainer, G. Grundmeier, M. Schlierf and A. Keller, Nanoscale, 2016, 8, 10398–10405 RSC.
- C. Kielar, F. v Reddavide, S. Tubbenhauer, M. Cui, X. Xu, G. Grundmeier, Y. Zhang and A. Keller, Angew. Chem., Int. Ed., 2018, 130, 15089–15093 CrossRef.
- J. B. Knudsen, L. Liu, A. L. B. Kodal, M. Madsen, Q. Li, J. Song, J. B. Woehrstein, S. F. J. Wickham, M. T. Strauss, F. Schueder, J. Vinther, A. Krissanaprasit, D. Gudnason, A. A. A. Smith, R. Ogaki, A. N. Zelikin, F. Besenbacher, V. Birkedal, P. Yin, W. M. Shih, R. Jungmann, M. Dong and K. v Gothelf, Nat. Nanotechnol., 2015, 10, 892–898 CrossRef CAS PubMed.
- K. Ebel and I. Bald, J. Phys. Chem. Lett., 2022, 4871–4876 CrossRef CAS PubMed.
- A. Keller, I. Bald, A. Rotaru, E. Cauët, K. v Gothelf and F. Besenbacher, ACS Nano, 2012, 6, 4392–4399 CrossRef CAS PubMed.
- L. Sala, H. Lyshchuk, J. Šáchová, D. Chvátil and J. Kočišek, J. Phys. Chem. Lett., 2022, 13, 3922–3928 CrossRef CAS PubMed.
- J. Rackwitz and I. Bald, Chem. – Eur. J., 2018, 24, 4680–4688 CrossRef CAS PubMed.
- L. Sala, A. Zerolová, A. Rodriguez, D. Reimitz, M. Davídková, K. Ebel, I. Bald and J. Kočišek, Nanoscale, 2021, 13, 11197–11203 RSC.
- S. le Caër, Water, 2011, 3, 235–253 CrossRef.
- S. M. Pimblott and J. A. LaVerne, Radiat. Phys. Chem., 2007, 76, 1244–1247 CrossRef CAS.
- E. Alizadeh, T. M. Orlando and L. Sanche, Annu. Rev. Phys. Chem., 2015, 66, 379–398 CrossRef CAS PubMed.
- E. Alizadeh and L. Sanche, Chem. Rev., 2012, 112, 5578–5602 CrossRef CAS PubMed.
- Y. Zheng and L. Sanche, Appl. Phys. Rev., 2018, 5, 021302 Search PubMed.
- A. Kumar, D. Becker, A. Adhikary and M. D. Sevilla, Int. J. Mol. Sci., 2019, 20, 3998 CrossRef CAS PubMed.
- J. Simons, Acc. Chem. Res., 2006, 39, 772–779 CrossRef CAS PubMed.
- R. Barrios, P. Skurski and J. Simons, J. Phys. Chem. B, 2002, 106, 7991–7994 CrossRef CAS.
- J. Narayanan S J, D. Tripathi and A. K. Dutta, J. Phys. Chem. Lett., 2021, 12, 10380–10387 CrossRef CAS PubMed.
- J. Spiegel, S. Adhikari and S. Balasubramanian, Trends Chem., 2020, 2, 123–136 CrossRef CAS PubMed.
- P. Wardman, Clin. Oncol., 2007, 19, 397–417 CrossRef CAS PubMed.
- L. Gong, Y. Zhang, C. Liu, M. Zhang and S. Han, Int. J. Nanomed., 2021, 16, 1083 CrossRef PubMed.
- R. Schürmann, S. Vogel, K. Ebel and I. Bald, Chem. – Eur. J., 2018, 24, 10271–10279 CrossRef PubMed.
- A. Keller, J. Rackwitz, E. Cauët, J. Liévin, T. Körzdörfer, A. Rotaru, K. v Gothelf, F. Besenbacher and I. Bald, Sci. Rep., 2014, 4, 1–6 Search PubMed.
- J. Rackwitz, M. L. Ranković, A. R. Milosavljević and I. Bald, Eur. Phys. J. D, 2017, 71, 1–9 CrossRef CAS.
- J. Rackwitz, J. Kopyra, I. Dąbkowska, K. Ebel, M. L. Ranković, A. R. Milosavljević and I. Bald, Angew. Chem., Int. Ed., 2016, 55, 10248–10252 CrossRef CAS PubMed.
- R. Schürmann, T. Tsering, K. Tanzer, S. Denifl, S. V. K. Kumar and I. Bald, Angew. Chem., Int. Ed., 2017, 56, 10952–10955 CrossRef PubMed.
- S. Vogel, K. Ebel, C. Heck, R. M. Schürmann, A. R. Milosavljević, A. Giuliani and I. Bald, Phys. Chem. Chem. Phys., 2019, 21, 1972–1979 RSC.
- S. Vogel, J. Rackwitz, R. Schürman, J. Prinz, A. R. Milosavljević, M. Réfrégiers, A. Giuliani and I. Bald, J. Phys. Chem. Lett., 2015, 6, 4589–4593 CrossRef CAS PubMed.
- S. Vogel, K. Ebel, R. M. Schürmann, C. Heck, T. Meiling, A. R. Milosavljevic, A. Giuliani and I. Bald, Chem. Phys. Chem., 2019, 20, 823–830 CrossRef CAS PubMed.
- T. Gerling and H. Dietz, Angew. Chem., Int. Ed., 2019, 58, 2680–2684 CrossRef CAS PubMed.
- N. P. Agarwal, M. Matthies, F. N. Gür, K. Osada and T. L. Schmidt, Angew. Chem., Int. Ed., 2017, 56, 5460–5464 CrossRef CAS PubMed.
- S. Ramakrishnan, L. Schärfen, K. Hunold, S. Fricke, G. Grundmeier, M. Schlierf, A. Keller and G. Krainer, Nanoscale, 2019, 11, 16270–16276 RSC.
- S. Ramakrishnan, H. Ijäs, V. Linko and A. Keller, Comput. Struct. Biotechnol. J., 2018, 16, 342–349 CrossRef CAS PubMed.
- G. Grossi, A. Jaekel, E. S. Andersen and B. Saccà, MRS Bull., 2017, 42, 920–924 CrossRef CAS.
- K. Kramm, T. Schröder, J. Gouge, A. M. Vera, K. Gupta, F. B. Heiss, T. Liedl, C. Engel, I. Berger, A. Vannini, P. Tinnefeld and D. Grohmann, Nat. Commun., 2020, 11, 1–12 CrossRef PubMed.
- C. Wang, K. Ebel, K. Heinze, U. Resch-Genger and I. Bald, Chem. – Eur. J., 2023, e202203719 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |