
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNDIPhos as a platform for chiral supramolecular ligands in rhodium-catalyzed enantioselective hydrogenation†
Guillaume
Force
a,
Robert J.
Mayer
 b,
Marie
Vayer
b and
David
Lebœuf
*b
b,
Marie
Vayer
b and
David
Lebœuf
*b
aInstitut de Chimie Moléculaire et des Matériaux d’Orsay (ICMMO), CNRS UMR 8182, Université Paris-Saclay, Orsay 91405, France
bInstitut de Science et d’Ingénierie Supramoléculaires (ISIS), CNRS UMR 7006, Université de Strasbourg, Strasbourg 67000, France. E-mail: dleboeuf@unistra.fr
First published on 26th April 2023
Abstract
Chiral naphthalene diimide ligands (NDIPhos) were exploited in rhodium-catalyzed enantioselective hydrogenation. The key feature of these ligands is their ability to self-assemble via π–π interactions to mimic bidentate ligands, offering a complementary method to traditional supramolecular strategies. This concept was further substantiated by computations with the composite electronic-structure method r2SCAN-3c.
Transition metal catalysis is instrumental to our daily life. It is indeed crucial to the production of high value-added molecules that are prevalent in our food, drugs, cosmetics, and agrochemicals. Two major breakthroughs in transition metal catalysis were related to the discovery of (1) bidentate ligands that form metal complexes of well-defined geometry through the chelation of the metal center for better control of the selectivity1 – currently the main players in catalysis – and (2) cationic organometallic complexes displaying weakly coordinating anions that readily ease the encounter with the substrates to accelerate transformations under milder reaction conditions.2 In search of new reactivity, synthetic chemists have designed hundreds of new bidentate ligands and thousands of new cationic complexes. However, these processes can become laborious as screening various catalysts can be hampered by limited access to libraries of structurally diverse ligands. This is particularly the case for bidentate ligands, whose preparation can be both time-consuming and expensive.
To overcome these drawbacks, the self-assembly of monodentate ligands via non-covalent interactions has emerged as a powerful strategy to mimic bidentate ligands around the metal center.3 This approach grants direct access to a large combinatorial library for catalysts through the straightforward preparation of monodentate ligands. Existing supramolecular approaches in transition metal asymmetric catalysis mostly rely on hydrogen bonding, coordination to a metal center, and electrostatic interactions to provide well-defined homo- and heterobidentate ligand–metal complexes as illustrated by the groups of Breit,4 Reek,5 Gennari,6 Nishibayashi,7 Takacs,8 van Leeuwen,9 Fan,10 and Vidal-Ferran (Fig. 1).11 However, in several cases, the incorporation of selective recognition units makes the synthesis of such assemblies more elaborate and costly than intended. Additionally, these non-covalent interactions can be disrupted by highly polar functional groups, which can drastically limit their efficiency and range of applications.
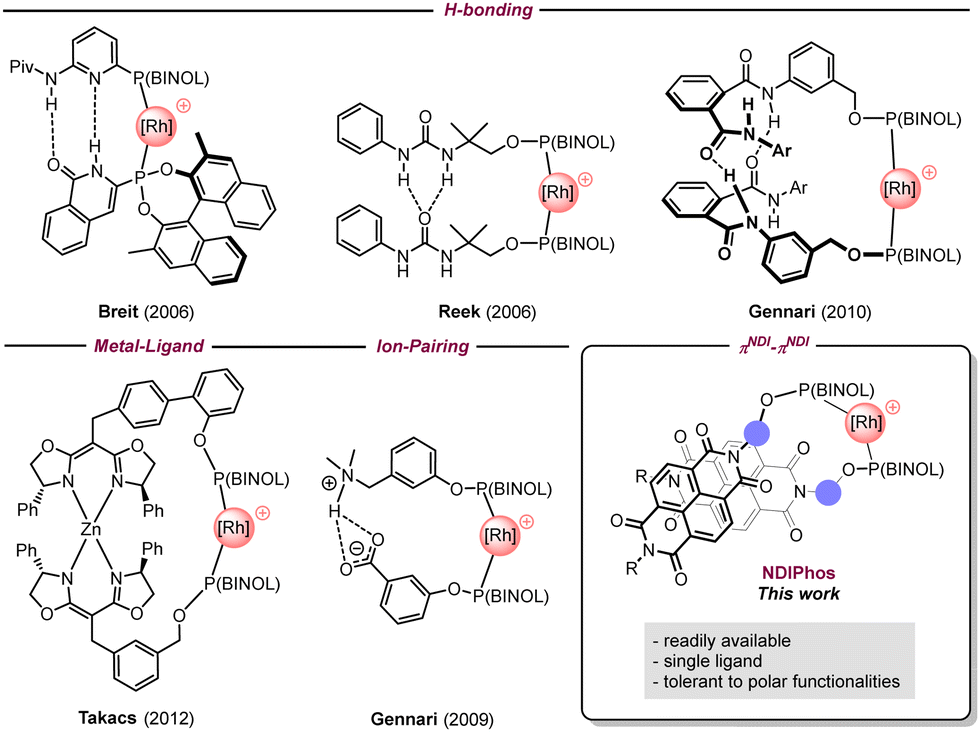 | ||
| Fig. 1 Selected examples of supramolecular self-assembled ligands for transition metal catalysis. | ||
In this context, we wondered if we could develop a supramolecular approach that retains the benefits of self-assembled ligands but eliminates their negative aspects. Hence, our attention was drawn to π–π interactions that, if sufficiently strong, are less prone to be impacted by polar functionalities. To date, the use of π-stacking for the design of self-assembled ligands remains underexplored in transition metal catalysis unless it is combined with other non-covalent interactions.12 Attempts have been made by the group of Gennari to use π–π interactions between perfluoro phenyls and electron-rich arenes,13 but without much success. The reason behind this result is likely the weakness of such interactions in solution. To overcome this issue, we assumed that naphthalene diimides (NDIs) that are well known for their ability to strongly self-assemble through π-stacking, could represent a viable solution to obtain stable formal bidentate ligands.14 While the use of NDIs has become increasingly popular in materials sciences, they have only been featured in a few applications in catalysis, mainly in π-anion catalysis by the group of Matile.15 Therefore, developing such an approach could expand the range of applications of NDIs in synthesis. Here we describe our efforts regarding the preparation of chiral self-assembled NDIPhos-based catalysts and their application to enantioselective hydrogenations as a proof of concept.
To validate our hypothesis regarding the capacity of NDI-derived ligands to undergo the anticipated self-assembly, we initially performed a computational analysis of the adduct formation of the model ligand L and Rh(COD)2+ by adapting a protocol from Grimme and co-workers for the study of non-covalent interactions (Fig. 2).16 This procedure relies on the composite electronic-structure method r2SCAN-3c, which includes the D4-dispersion correction and a geometrical counterpoise correction.17 It was shown that this functional achieves comparable results to classical hybrid-DFT methods, but only with a fraction of the computational cost, making it particularly suitable for large systems like ours. After a conformational search at the GFN2-xTB(ALPB = CH2Cl2) level with the CREST program,18 structures were optimized at the r2SCAN-3c(SMD = CH2Cl2) level of theory within ORCA.19,20 Thermochemical corrections were obtained using the single-point Hessian (SPH) approach at the GFN2-xTB(ALPB = CH2Cl2) level with the r2SCAN-3c(SMD = CH2Cl2)-optimized structures as input. On the energetically lowest conformers obtained in this way, additional numerical frequency calculations were performed using the r2SCAN-3c(SMD = CH2Cl2) method.21 For further verification of the methods, control computations were performed at the (SMD = CH2Cl2)/M06-L/def2-TZVP level. As evidenced by an analysis of the non-covalent interactions (NCIs),22 the model ligand L already features intramolecular π-stacking interactions between the NDI moiety and the naphthyl group. Dimerization of L to (L)2 was computed to be exergonic at the r2SCAN-3c(SMD = CH2Cl2) level (ΔG = −10.0/−15.6 kJ mol−1) but slightly endergonic at the (SMD = CH2Cl2)/M06-L/def2-TZVP level (ΔG = +9.4 kJ mol−1). The resulting structure features extensive π–π and CH–π interactions between the NDI and the naphthyl units. In (L)2, both phosphorus atoms are oriented in a way that allows (L)2 to act as a bidentate ligand to yield a cis-phosphite rhodium complex. The binding of Rh(COD)+ at (L)2 was computed to be highly favorable with all methods (ΔG = −81.8/−90.6/−98.7 kJ mol−1) and results in only insignificant structural changes of the (L)2 fragment, thereby retaining the intramolecular interactions, notably the π-stacking. In contrast, the binding of Rh(cod)+ at the ligand L itself is computed to be less favored (ΔG = −7.4/+ 0.2/−22.8 kJ mol−1). While one carbonyl group of the NDI allows L to act as a bidentate ligand along with the phosphorus atom, this ligand is unlikely a good ligand to favor the formation of such a complex. The weak binding of L with Rh(cod)+ is not surprising as only minor intramolecular interactions between the COD ligand and one of the aryl rings of L are retained in the Rh(COD)(L)+ adduct. In parallel, we recorded the 31P NMR spectra of the mixture of [Rh(COD)2]OTf and L1 (ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and ratio 1:1) in CD2Cl2. Here, the methyl group of L was replaced by an n-pentyl group to improve the solubility of the corresponding ligand. Both spectra display a doublet at 120.5 ppm with 1JP,Rh = 259.4 Hz, which is consistent with the two phosphites coordinating to the rhodium center in a cis-fashion (see ESI† for details), while the formation of [Rh(COD)L1]OTf was not observed.
:2 and ratio 1:1) in CD2Cl2. Here, the methyl group of L was replaced by an n-pentyl group to improve the solubility of the corresponding ligand. Both spectra display a doublet at 120.5 ppm with 1JP,Rh = 259.4 Hz, which is consistent with the two phosphites coordinating to the rhodium center in a cis-fashion (see ESI† for details), while the formation of [Rh(COD)L1]OTf was not observed.
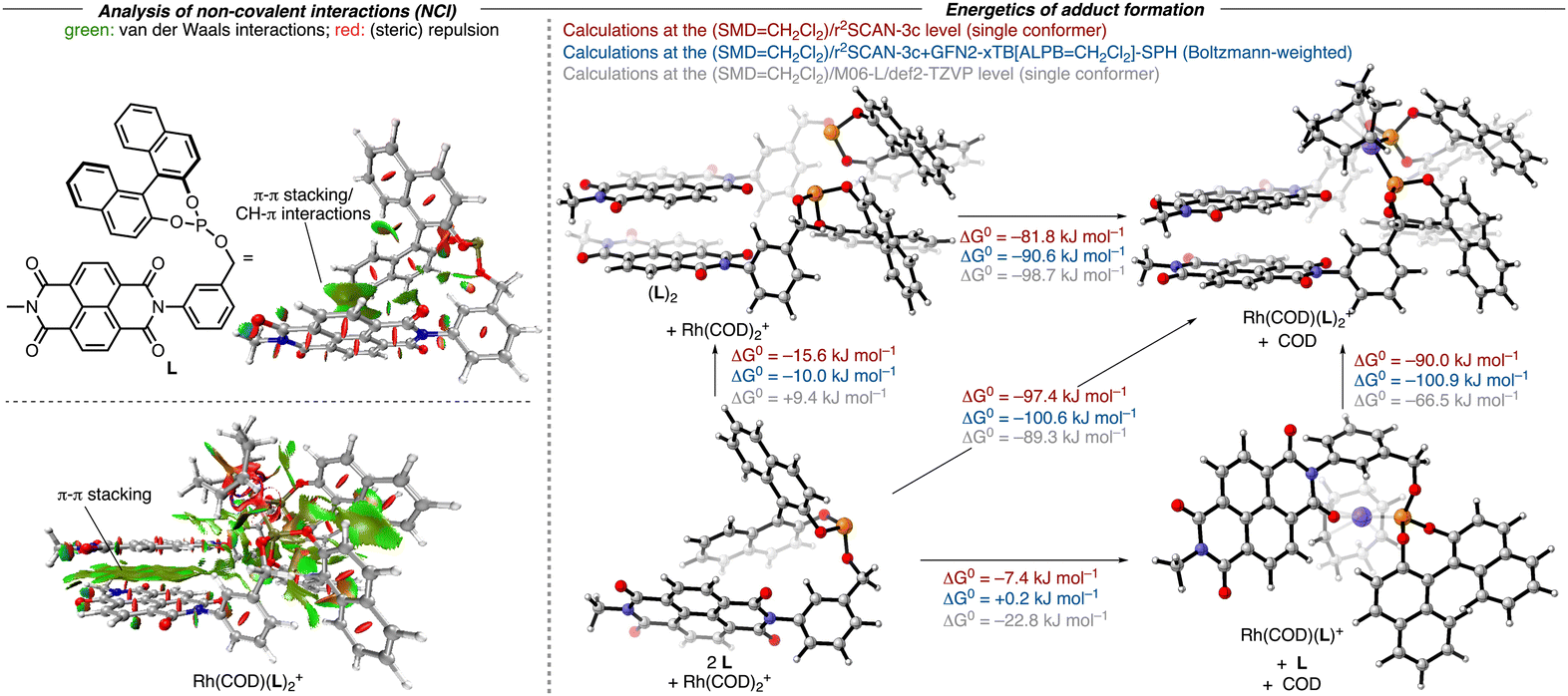 | ||
| Fig. 2 Left: NCI analysis of the structures at the (SMD = CH2Cl2)/r2SCAN-3c level. Right: Geometries of the most favorable conformers for each species optimized at the (SMD = CH2Cl2)/r2SCAN-3c and (SMD = CH2Cl2)/M06-L/def2-TZVP levels and associated Gibbs free energies for complex formation calculated in three different ways. | ||
Encouraged by the computational analysis that indicates the defined binding of Rh+ in the pre-associated (L)2 system, we started to evaluate the potential of chiral ligand L1 in the Rh-catalyzed enantioselective hydrogenation of methyl (Z)-2-acetamido-3-phenylacrylate at room temperature under 20 atm of H2 for 24 h (Table 1). Using [Rh(COD)(MeCN)2]BF4 as a rhodium source (1 mol%), we first investigated the influence of the solvent on the reactivity (entries 1–9). Except for nitromethane, all rhodium complexes were catalytically active with the enantiomeric excesses ranging from 9 to 93%. Here, the best result was obtained with nitrobenzene as a solvent. Interestingly, such a positive effect of nitrobenzene on the reactivity of NDI was already observed by the group of Matile in enantioselective π-anion catalysis.15c Based on their observations, a plausible explanation is that an electron-deficient arene might increase the electron-deficiency of the NDI, reinforcing the π–π interaction between the NDI units. Of note, the amount of nitrobenzene could be reduced by using a mixture of CH2Cl2/PhNO2 (10:1), affording the product with 95% ee (entry 9). Other cationic rhodium sources were also tested and overall produced comparable results (entries 10–13). Reducing the reaction temperature to 0 °C allowed us to slightly improve the enantiomeric excess to 97% (entry 12).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Conversionb (%) | eec (%) abs. config.d |
|
a Reaction conditions: substrate/ligand/catalyst = 100:2:1, solvent c = 0.13 M, and T = 25 °C.
b Determined by 1H NMR.
c Determined by HPLC using a chiral column Daicel Chiralpak IA.
d Assignment based on comparison with ref. 21.
e Reaction at 0 °C.
|
||||
| 1 | [Rh(cod)(MeCN)2]BF4 | CH2Cl2 | >99 | 87, S |
| 2 | [Rh(cod)(MeCN)2]BF4 | Toluene | >99 | 71, S |
| 3 | [Rh(cod)(MeCN)2]BF4 | Benzene | >99 | 80, S |
| 4 | [Rh(cod)(MeCN)2]BF4 | PhNO2 | >99 | 93, S |
| 5 | [Rh(cod)(MeCN)2]BF4 | PhBr | >99 | 92, S |
| 6 | [Rh(cod)(MeCN)2]BF4 | 1,4-Dioxane | >99 | 35, S |
| 7 | [Rh(cod)(MeCN)2]BF4 | MeNO2 | <5 | — |
| 8 | [Rh(cod)(MeCN)2]BF4 | MeOH | >99 | 9, S |
| 9 | [Rh(cod)(MeCN)2]BF4 | CH2Cl2/PhNO2 (10:1) |
>99 | 95, S |
| 10 | [Rh(COD)2]OTf | CH2Cl2/PhNO2 (10:1) |
>99 | 96, S |
| 11 | [Rh(COD)2]SbF6 | CH2Cl2/PhNO2 (10:1) |
>99 | 96, S |
| 12e | [Rh(COD)2]SbF6 | CH2Cl2/PhNO2 (10:1) |
>99 | 97, S |
| 13 | [Rh(COD)2]BARF | CH2Cl2/PhNO2 (10:1) |
>99 | 96, S |
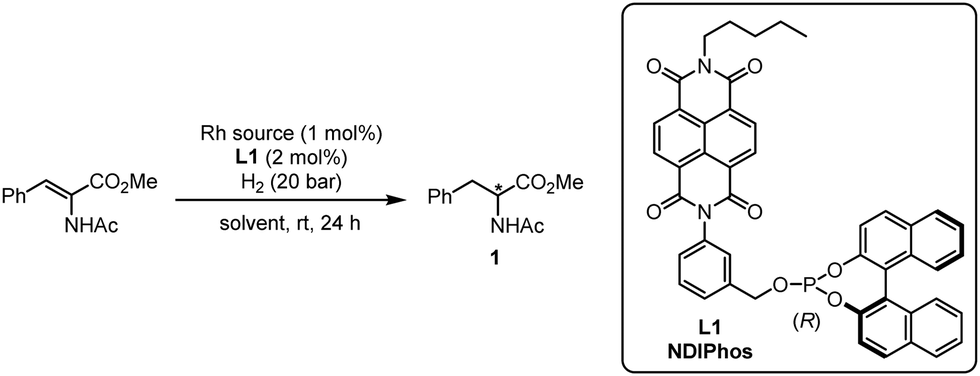
We then compared the reactivity and efficiency of L1 with other ligands incorporating electron-deficient aromatic scaffolds (Fig. 3). In particular, ligand L2, which has a similar structure to the BenzaPhos ligands developed by Gennari,23 provided the highest enantiomeric excess (ee 98%). Tetrafluorophthalimido-derived ligand L3 also proved efficient in the transformation (ee 96%). In this case, analogous computations still indicated a well-defined behavior of a pre-associated bidentate ligand for rhodium. However, this pre-association is energetically less favored and not as defined as with L1 (see Fig. S2 for details, ESI†). Of note, switching the position of the phosphite moiety from the meta to para position in L1 (L6) led to a slight decrease of the enantiomeric excess (ee 91%). In the same vein, a drop of the enantiomeric excess (ee 81%) was observed by removing fluorine from L3, which might be attributed to the fact that the π-stacking is not possible anymore. Other ligands were also tested but did not afford enantiomeric excesses superior to 90%.
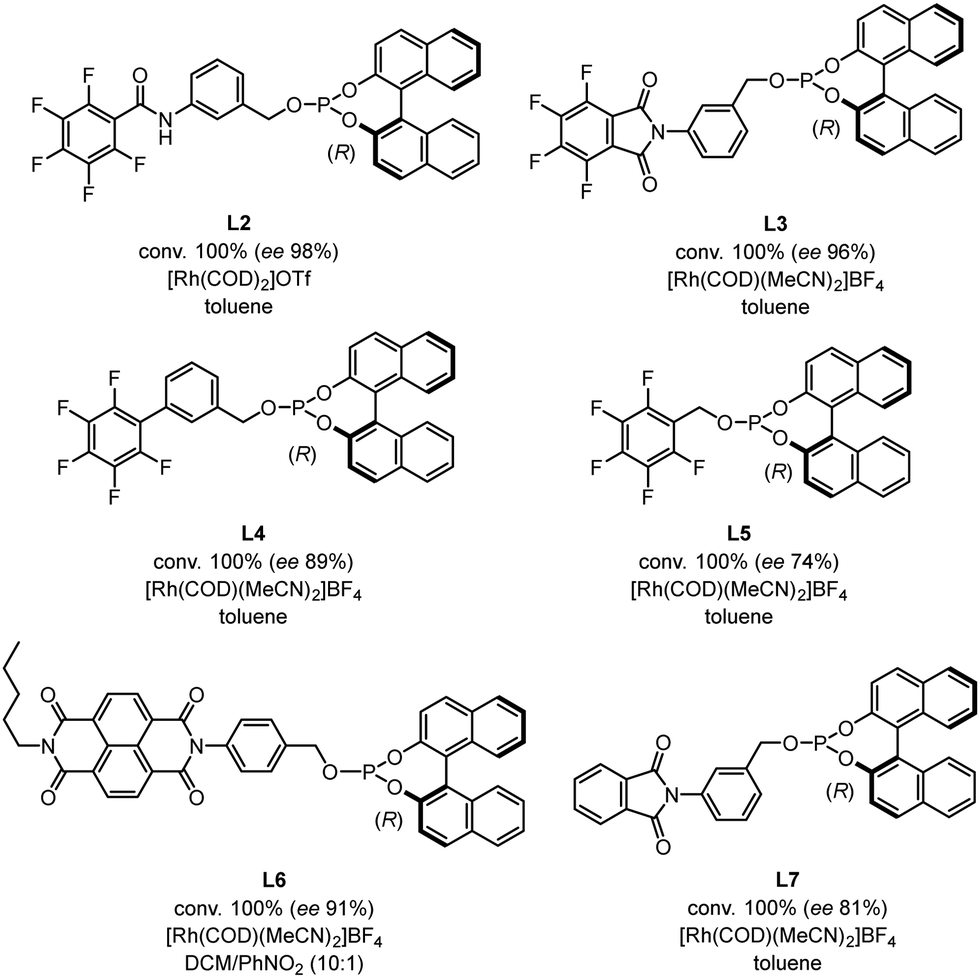 | ||
| Fig. 3 Comparison with other electron-deficient ligands. | ||
Lastly, we explored the synthetic potential of L1 with various substrates (Fig. 4), using L2 and L3 as references. In the case of classic substrates such as methyl 2-acetamidoacrylate, dimethyl itaconate, and N-(3,4-dihydronaphthalen-1-yl)acetamide, all the ligands provided fairly similar results for products 2–4. On the other hand, in the case of 2-acetamidoacrylic acid, we observed significant differences as L1 afforded 89% ee for 5, while L2 gave 5% ee, and L3 gave 37% ee. This clearly demonstrates that in the case of a ligand prone to H-bonding coordination such as L2, the presence of a highly polar functional group such as a carboxylic acid is detrimental to the selectivity of the reaction. In addition, if the π-stacking interaction is not strong enough, the reaction becomes less selective as shown by L3. L1 also proved highly selective when a spacer was added (6) and when a tetrasubstituted substrate was used (7).
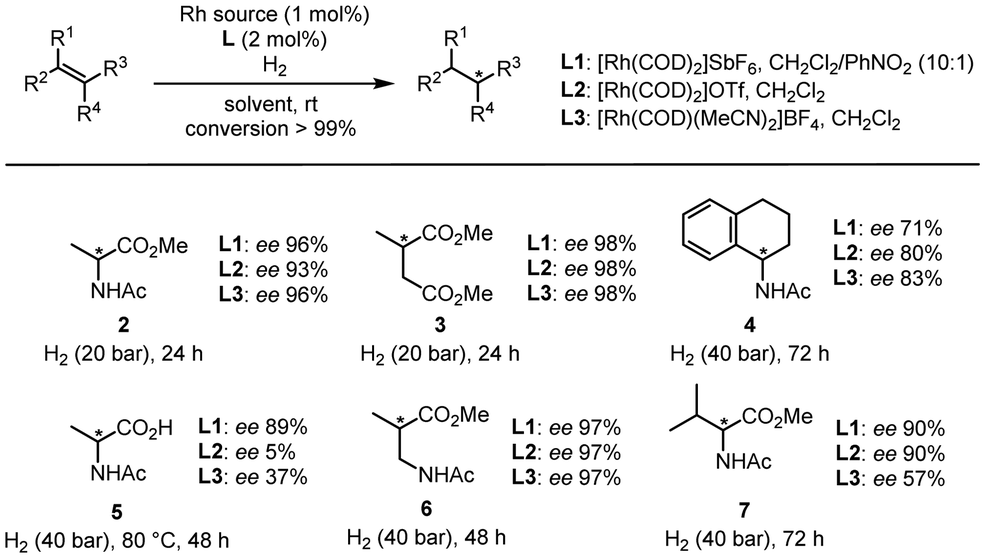 | ||
| Fig. 4 Scope for the rhodium-catalyzed enantioselective hydrogenation. | ||
In conclusion, we described a new class of self-assembled bidentate NDI-derived ligands featuring π–π interactions, which were successfully used in rhodium-catalyzed enantioselective hydrogenation of diverse substrates. The catalysts incorporating this NDIPhos ligand were generally highly selective, notably in the presence of polar functionalities such as carboxylic acids. Our current efforts are dedicated to the improvement of their design to expand their range of applications.
This work was supported by the ANR (ANR-16-CE07-0022, funding for G. F.) and the Interdisciplinary Thematic Institute ITI-CSC via the IdEx Unistra (ANR-10-IDEX-0002, funding for M. V.) within the program Investissement d’Avenir. R. J. M. thanks the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) for the fellowship (MA 9687/1-1). Computations were performed at the High Performance Computing Center of the University of Strasbourg. Part of the computing resources was funded by the Equipex Equip@Meso project (Programme Investissements d'Avenir) and the CPER Alsacalcul/Big Data. D. L. thanks the CNRS. D. L. thanks Pr. Vincent Gandon and Dr Emmanuelle Schulz for useful discussions. The authors thank Emilie Kolodziej for her help with the HPLC and GC analyses.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) T. P. Dang and H. B. Kagan, J. Chem. Soc. D, 1971, 481 RSC; (b) W. S. Knowles, M. J. Sabacky, B. D. Vineyard and D. J. Weinkauff, J. Am. Chem. Soc., 1975, 97, 2567 CrossRef CAS; (c) A. Miyashita, A. Yasuda, H. Takaya, K. Toriumi, T. Ito, T. Souchi and R. Noyori, J. Am. Chem. Soc., 1980, 102, 7932 CrossRef CAS.
- (a) J. A. Osborn and R. R. Schrock, J. Am. Chem. Soc., 1971, 93, 2397 CrossRef CAS; (b) J. A. Osborn and R. R. Schrock, J. Am. Chem. Soc., 1976, 98, 4450 CrossRef.
- For selected reviews on supramolecular catalysis, see: (a) J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615 CrossRef CAS PubMed; (b) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660 RSC; (c) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012 CrossRef CAS PubMed; (d) J. N. H. Reek, B. de Bruin, S. Pullen, T. J. Mooibroek, A. M. Kluwer and X. Caumes, Chem. Rev., 2022, 22, 12308 CrossRef PubMed; (e) Supramolecular Catalysis, ed. P. W. N. M. van Leeuwen and M. Raynal, Wiley-VCH, Weinheim, 2022 Search PubMed.
- (a) M. Weis, C. Waloch, W. Seiche and B. Breit, J. Am. Chem. Soc., 2006, 128, 4188 CrossRef CAS PubMed; (b) A. C. Laungani, J. M. Slaterry, I. Krossing and B. Breit, Chem. – Eur. J., 2008, 14, 4488 CrossRef CAS PubMed; (c) J. Wieland and B. Breit, Nat. Chem., 2010, 2, 832 CrossRef CAS PubMed.
- (a) X.-B. Jiang, L. Lefort, P. E. Goudriaan, A. H. M. de Vries, P. W. N. M. van Leeuwen, J. G. de Vries and J. N. H. Reek, Angew. Chem., Int. Ed., 2006, 45, 1223 CrossRef CAS PubMed; (b) A. J. Sandee, A. M. van der Burg and J. N. H. Reek, Chem. Commun., 2007, 864 RSC; (c) P.-A. R. Breuil, F. W. Patureau and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 2162 CrossRef CAS PubMed.
- (a) L. Pignataro, S. Carboni, M. Civera, R. Colombo, U. Piarulli and C. Gennari, Angew. Chem., Int. Ed., 2010, 49, 6633 CrossRef CAS PubMed; (b) L. Pignataro, B. Lynikaite, J. Cvengroš, M. Marchini, U. Piarulli and C. Gennari, Eur. J. Org. Chem., 2009, 2539 CrossRef CAS.
- G. Hattori, T. Hori, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2007, 129, 12930 CrossRef CAS PubMed.
- N. C. Thacker, S. A. Moteki and J. M. Takacs, ACS Catal., 2012, 2, 2743 CrossRef CAS PubMed.
- P. W. N. M. van Leeuwen, D. Rivillo, M. Raynal and Z. Freixa, J. Am. Chem. Soc., 2011, 133, 18562 CrossRef CAS PubMed.
- Y. Li, B. Ma, Y. He, F. Zhang and Q.-H. Fan, Chem. – Asian J., 2010, 5, 2454 CrossRef CAS PubMed.
- I. Mon, D. A. Jose and A. Vidal-Ferran, Chem. – Eur. J., 2013, 19, 2720 CrossRef CAS PubMed.
- (a) A. C. Laugani and B. Breit, Chem. Commun., 2008, 844 RSC; (b) M. Raynal, F. Portier, P. W. N. M. van Leeuwen and L. Bouteiller, J. Am. Chem. Soc., 2013, 135, 17687 CrossRef CAS PubMed.
- B. Lynikaite, J. Cvengroš, U. Piarulli and C. Gennari, Tetrahedron Lett., 2008, 49, 755 CrossRef CAS.
- For recent reviews, see: (a) M. Al Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 11685 CrossRef PubMed; (b) Y. Zhao, Y. Cotelle, L. Liu, J. Lopez-Andarias, A.-B. Bornhof, M. Akamatsu, N. Sakai and S. Matile, Acc. Chem. Res., 2018, 51, 2255 CrossRef CAS PubMed; (c) S. V. Bhosale, M. Al Kobaisi, R. W. Jadhav, P. P. Morajkar, L. A. Jones and S. George, Chem. Soc. Rev., 2021, 50, 9845 RSC.
- For selected examples, see: (a) Y. Zhao, Y. Cotelle, A.-J. Avestro, N. Sakai and S. Matile, J. Am. Chem. Soc., 2015, 137, 11582 CrossRef CAS PubMed; (b) Y. Cotelle, S. Benz, A.-J. Avestro, T. R. Ward, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2016, 55, 4275 CrossRef CAS PubMed; (c) L. Liu, Y. Cotelle, A.-J. Avestro, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 7876 CrossRef CAS PubMed; (d) L. Liu, Y. Cotelle, J. Klehr, N. Sakai, T. R. Ward and S. Matile, Chem. Sci., 2017, 8, 3770 RSC; (e) L. Liu, Y. Cotelle, A.-B. Bornhof, C. Besnard, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2017, 56, 13066 CrossRef CAS PubMed; (f) M. Paraja and S. Matile, Angew. Chem., Int. Ed., 2020, 59, 6273 CrossRef CAS PubMed.
- (a) M. Bursch, J.-M. Mewes, A. Hansen and S. Grimme, Angew. Chem., Int. Ed., 2022, 61, e202205735 CrossRef CAS PubMed; (b) J. Gorges, S. Grimme and A. Hansen, Phys. Chem. Chem. Phys., 2022, 24, 28831–28843 RSC.
- S. Grimme, A. Hansen, S. Ehlert and J.-M. Mewes, J. Chem. Phys., 2021, 154, 064103 CrossRef CAS PubMed.
- (a) P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169 RSC; (b) C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652 CrossRef CAS PubMed; (c) S. Ehlert, M. Stahn, S. Spicher and S. Grimme, J. Chem. Theory Comput., 2021, 17, 4250 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, e1606 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2013, 9, 609 CrossRef CAS PubMed.
- S. Spicher and S. Grimme, J. Chem. Theory Comput., 2021, 17, 1701 CrossRef CAS PubMed.
- (a) E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS PubMed; (b) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- L. Pignataro, C. Bovio, M. Civera, U. Piarulli and C. Gennari, Chem. – Eur. J., 2012, 18, 10368 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc00695f |
| This journal is © The Royal Society of Chemistry 2023 |