
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revealing the nitrogen reaction pathway for the catalytic oxidative denitrification of fuels†
Michael
Huber
 a,
Maximilian J.
Poller
a,
Jens
Tochtermann
b,
Wolfgang
Korth
b,
Andreas
Jess
b and
Jakob
Albert
*a
a,
Maximilian J.
Poller
a,
Jens
Tochtermann
b,
Wolfgang
Korth
b,
Andreas
Jess
b and
Jakob
Albert
*a
aInstitute of Technical and Macromolecular Chemistry, Universität Hamburg, Bundesstraße 45, 20146 Hamburg, Germany. E-mail: jakob.albert@uni-hamburg.de
bhair of Chemical Engineering, Center of Energy Technology (ZET), University of Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany
First published on 14th March 2023
Abstract
Aside from the desulfurisation, the denitrogenation of fuels is of great importance to minimze the environmental impact of transport emissions. The oxidative reaction pathway of organic nitrogen in the catalytic oxidative denitrogenation could be successfully elucidated. This is the first time such a pathway could be traced in detail in non-microbial systems. It was found that the organic nitrogen is first oxidized to nitrate, which is subsequently reduced to molecular nitrogen via nitrous oxide. Hereby, the organic substrate serves as a reducing agent. The discovery of this pathway is an important milestone for the further development of fuel denitrogenation technologies.
The United Nations aim to counteract global warming with Net Zero Emission (NZE) commitments; however, it is not yet foreseeable when crude oil-based fuels will become obsolete. In 2021, more than 50 million barrels per day (mb d−1) were consumed for the transport sector alone. And if the NZE goes according to plan, only 3 mb d−1 will be saved for road freight in 2030. The Stated Policies Scenario (STEPS) sees the development somewhat more conservatively; according to its projection consumption is even 1.5 mb d−1 more in road freight.1 Therefore, it is even more important to make clean fuels available in order to minimize environmental impact. Above all, heteroatoms such as sulfur or nitrogen produce SO2 and NOx during combustion in the engines, which is not only harmful to the climate but also to health.2–4 Therefore, in refineries, these heteroatoms are removed by hydrotreating to produce clean fuels.5 However, this catalytic reaction is inhibited by the basic, nitrogenous reactants (e.g., quinoline) as well as by NH3. The ion pair of the nitrogen atom forms strong pi-bonds to the active sites of the hydrotreating catalyst, which diminishes its activity.5 This effect can already be observed from a nitrogen concentration of 5 ppm and is further intensified when the inhibitory reactants occur in combination.6 For this reason, it is important to limit the nitrogen content of the product oil to below 10 ppmw.7
There is a variety of research into alternatives to hydrotreating, including extraction,8–12 selective adsorption,13–17 and oxidation18–22 approaches. To maximize the desulfurization and denitrogenation effectiveness in comparison to just extraction and adsorption, selective oxidation is typically combined with either extraction or selective adsorption. The selective oxidation produces more polar compounds that can be removed from the non-polar oil in a separate step.20,22,23 The extraction step can also be carried out in parallel to the oxidation reaction, as a result of in situ separation of the oxidation products (ECODS; extractive catalytic oxidative desulfurization). In this process, H8PV5Mo7O40 (HPA-5) is employed as a homogeneous polyoxometalate (POM) catalyst in an aqueous phase, whereas the fuel forms a separate organic liquid phase. The sulfur containing fuel components are oxidized after diffusion from the organic fuel phase into the aqueous catalyst phase, to form highly polar products such as H2SO4 and carboxylic acids, which are thereby extracted from the organic fuel phase and accumulate in the aqueous phase.24–27 In contrast to the inhibiting properties of the basic nitrogen compounds in hydrotreating, the oxidative desulfurization improves with simultaneous denitrification in this system (ECODN; extractive catalytic oxidative denitrogenation).26 The reaction pathway of ECODS has already been well studied.25,26,28 In contrast, the oxidation of nitrogen compounds in ECODN is not yet well understood and requires more detailed investigations.
In principle, the ECODN process can be performed just like the ECODS process: the substrate is oxidized using HPA-5 and molecular oxygen in an additional aqueous phase. Thereby, the carbon of the substrate can be fully oxidized to carbon dioxide and carboxylic acids like formic or acetic acid as in ECODS.24
This leads to the question, whether the oxidation of nitrogen atoms leads to nitrate, as sulfur ends up in sulfate ions, or does the reaction yield molecular nitrogen (N2).28 In order to answer this question and to elucidate the nitrogen reaction pathway in the ECODN process, we have first closed the nitrogen balance and subsequently investigated the decomposition pathway of nitrogen containing molecules under ECODN conditions.
The ubiquity of nitrogen makes the detection of molecular nitrogen as a reaction product, and thereby closing the nitrogen balance of the reaction, particularly challenging. All investigations must be carried out in the absence of air. Therefore, all solvents used were degassed with argon. After the initial aqueous phase was added to the reactor, the gas phase was flushed three times with 30 bar oxygen while stirring vigorously with a gas entrainment impeller. Subsequently, the reactor was heated several times with repeated exchanges of the gas phase. Thereby the nitrogen content in the reactor was decreased to 0.07 mol% of the gaseous phase, which corresponds to an absolute amount of 0.16 mmol.
The reaction was started by adding the reactant (quinaldine in iso-octane (0.21 mol L−1)) in portions up to a total amount of 4.6 mmol quinaldine. After 73 h the reaction was terminated by cooling down to room temperature. At this point, complete conversion of quinaldine was achieved as indicated by gas chromatography-mass spectrometry (GC-MS). Aside from the two liquid phases and the gaseous phase, the reactor contained some solid residue, which was analyzed by CHNSO elemental analysis (details are provided in ESI†) and determined to 5,8-quinaldinedione (Table S1, ESI†). The aqueous phase was analyzed by High Performance Liquid Chromatography (HPLC) and contains 10% acetic acid, 9% acetone and 7% formic acid, respectively, adding to the carbon balance (percentages are based on amount of carbon in initial substrate). The gas phase was analyzed by micro gas chromatography (Micro-GC) and shows 63% CO2, as well as 2% CO. Another 11% of the carbon is found in the insoluble precipitate. Overall, 93% (2.2 mmol) of the nitrogen atoms form molecular nitrogen, the rest can be found in the oxidized solid residue (Fig. 1). Since both carbon and nitrogen mass balance can be closed, it can be concluded that all nitrogen from the substrate would turn into N2 by oxidation.
 | ||
| Fig. 1 Product composition of a long-term experiment for the conversion of quinaldine to detect the nitrogen reaction product. Reaction conditions: 4.6 mmol quinaldine in 20 mL iso-octane, 0.2 mmol of HPA 5 in 200 mL water, 120 °C, 20 bar O2, 1000 rpm for 73 h. | ||
However, this does not exclude also nitrate formation as an intermediate. In microbial processes, a reaction pathway is described in which bacteria form nitrate from nitrogen containing compounds under oxidative conditions, which is then reduced to N2via N2O.29–32 In classic chemistry, too, reducing NOx to N2 under oxidative conditions is possible.33 Here, organic molecules such as methane or propene serve as reducing agent.33,34 When converting quinaldine, the molecule itself could be a reduction agent for nitrate and its intermediates.
We assume that organic nitrogen like quinaldine is first oxidized to nitrate and then reduced to N2via N2O (Fig. 2).
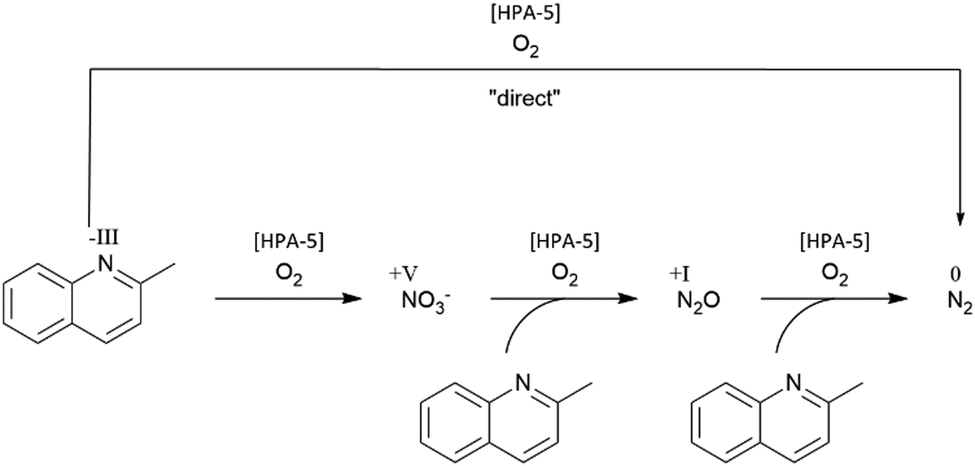 | ||
| Fig. 2 Reaction pathway from nitrogen with the “direct” route to N2 and the “indirect” route via nitrate and nitrous oxide to N2. | ||
In order to clarify whether the reaction pathway proceeds directly to N2 (Fig. 2, “direct”) or whether other NOx intermediates are formed (Fig. 2, “indirect”), we conducted further investigations using nitrate as a substrate. Hence, 100 mmol (42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 671 mg L−1) sodium nitrate in iso-octane as a reactant were added to our reaction system together with the aqueous HPA-5 catalyst solution. This mixture was processed under typical ECODN conditions (120 °C, 20 bar O2). The experiment showed no conversion of nitrate. The loss of <2% nitrate was due to the fact that a prior separation of the catalyst was required for the measurement by ion chromatography (IC) (see Table 1). This was done by nanofiltration through a membrane system. It was found that no nitrate was converted during the reaction. This was quite expected since the reducing agent for the reduction of nitrate under these strong oxidation conditions was absent.
671 mg L−1) sodium nitrate in iso-octane as a reactant were added to our reaction system together with the aqueous HPA-5 catalyst solution. This mixture was processed under typical ECODN conditions (120 °C, 20 bar O2). The experiment showed no conversion of nitrate. The loss of <2% nitrate was due to the fact that a prior separation of the catalyst was required for the measurement by ion chromatography (IC) (see Table 1). This was done by nanofiltration through a membrane system. It was found that no nitrate was converted during the reaction. This was quite expected since the reducing agent for the reduction of nitrate under these strong oxidation conditions was absent.
Therefore, the experiment was repeated using a modified method. Hereby, 3 mmol thiophene (13856 mg L−1) was used as a reducing agent, and oxygen was replaced by nitrogen. Thiophene was chosen as a reducing agent because a nitrogen containing compound might bias the measurement results. Generally, the use of a hydrocarbon as reducing agent was inspired by J. N. Armor et al.33–35 Again, no conversion of nitrate (IC) nor thiophene (GC-MS) was observed (see Table 1). The fact that thiophene was not converted indicates that nitrate cannot substitute O2 as an oxidant. Moreover, it can be assumed that oxygen is required to start the reduction of nitrate, as it is necessary for the reduction of NOx. This fact also fits the results of previous research by J. N. Armor et al.33–35
Finally, the reaction of nitrate with thiophene was studied under oxidative conditions (O2 atmosphere). This lead to a conversion of 32% of nitrate, while 95% of thiophen was also oxidized to classical ECODS products like sulfuric acid, formic acid and acetic acid (see Table 1). In addition, N2O could also be detected qualitatively with the Micro-GC, which corresponds to the microbial degradation path. This indicates that the indirect reaction pathway via the formation of nitrate is possible in this reaction system, whereby a heteroaromatic substrate is needed as a reducing agent, whereas the main fuel does not react.
In previous experiments, nitrate or nitrous oxide could never be detected, so it can be assumed that these molecules are converted very fast and do not accumulate in the reaction mixture. In order to check whether they are formed at all during the conversion of quinaldine and further converted to N2via the indirect reaction pathway, the reactor setup had to be adjusted. Therefore, online gas phase sampling was set up in a way that samples could be taken every 5 min from the bypass stream and analyzed by Micro-GC. This is to prove if the reaction produces nitrous oxide. However, since it can be assumed that the nitrous oxide is a very reactive intermediate and only occurs in very low concentrations that cannot be detected with the Micro-GC, an additional cold trap was installed in the exhaust gas flow of the bypass after the Micro-GC. The cold trap was cooled to approx. −130 °C with a cold mixture of n-pentane and liquid nitrogen.36 Therefore, the melting point of nitrous oxide (m. p. −91 °C)37 was significantly undershot in order to freeze it without enriching oxygen (b. p. −183 °C).38 After the reaction, the cold trap was thawed and the gas phase was measured by GC-MS. In addition, part of the aqueous phase was removed after six hours to prevent further reaction of the nitrate formed. The HPA-5 catalyst was removed by membrane separation39 and the permeate was measured by IC.
The test for detecting nitrate or nitrous oxide was carried out using 4 mmol of quinaldine. In the aqueous phase, 0.42 mg L−1 nitrate could be detected by IC. However, the Micro-GC in this setup was not sensitive enough for the clear evidence of N2O. Therefore, further investigations became necessary. To verify the detection of nitrate, the experiment was repeated with 4 mmol of quinoline as a substrate. Samples were taken from the aqueous phase after five, six and seven hours, respectively. The sample mixture was separated in the membrane system and then analyzed for nitrate and nitrite by IC. 2.77 mg L−1 nitrate were detected, but no nitrite. This proved that the oxidative conversion of nitrogen compounds in this system takes place via the indirect reaction pathway.
The fact that NO3− is not accumulated in the aqueous phase in significant amounts indicates, that the decomposition of NO3− to N2O proceeds faster than the initial oxidation of the organic substrate to NO3−. We furthermore conclude that the reaction of N2O to N2 is also very fast, as no accumulation of N2O was observed at all.
In summary, we were able to prove without doubt that N2 is the final product of oxidative denitrogenation of nitrogen-containing model gasoline. Additionally, it was shown that the degradation pathway can proceed via two different ways, with the indirect route being clearly demonstrated by a series of experiments. This is the first time this degradation pathway of organic nitrogen compounds via nitrate has been proven in non-microbial systems under oxidative conditions.
The authors would like to thank the Deutsche Forschungsgemeinschaft (DFG, German research foundation) for financial support through their projects JE 257/20-1 and AL 2130/3-2.
Conflicts of interest
There are no conflicts to declare.Notes and references
- IEA, World Energy Outlook 2022, International Energy Agency, Paris, 2022 Search PubMed.
- A. Zecca and R. S. Brusa, Il Nuovo Cimento C, 1992, 15, 481–483 CrossRef.
- G. Lammel and H. Grassl, Environ. Sci. Pollut. Res., 1995, 2, 40–45 CrossRef CAS PubMed.
- C. Pénard-Morand and I. Annesi-Maesano, Breathe, 2004, 1, 108–119 CrossRef.
- C. S. Hsu and P. R. Robinson, Springer Handbook of Petroleum Technology, Springer, 2017 Search PubMed.
- G. C. Laredo S, J. A. De Los Reyes H, J. Luis Cano D and J. Jesús Castillo M, Appl. Catal., A, 2001, 207, 103–112 CrossRef CAS.
- J. G. Speight, The Chemistry and Technology of Petroleum, CRC Press, 5 edn, 2006 Search PubMed.
- A. Bosmann, L. Datsevich, A. Jess, A. Lauter, C. Schmitz and P. Wasserscheid, Chem. Commun., 2001, 2494–2495 RSC.
- J. Eßer, P. Wasserscheid and A. Jess, Green Chem., 2004, 6, 316–322 RSC.
- P. S. Kulkarni and C. A. M. Afonso, Green Chem., 2010, 12, 1139–1149 RSC.
- S. Zhang, Q. Zhang and Z. C. Zhang, Ind. Eng. Chem. Res., 2004, 43, 614–622 CrossRef CAS.
- J. Zhang, J. Xu, J. Qian and L. Liu, Pet. Sci. Technol., 2013, 31, 777–782 CrossRef CAS.
- J. H. Kim, X. Ma, A. Zhou and C. Song, Catal. Today, 2006, 111, 74–83 CrossRef CAS.
- J. M. Kwon, J. H. Moon, Y. S. Bae, D. G. Lee, H. C. Sohn and C. H. Lee, ChemSusChem, 2008, 1, 307–309 CrossRef CAS.
- D. Liu, J. Gui and Z. Sun, J. Mol. Catal. A: Chem., 2008, 291, 17–21 CrossRef CAS.
- S. Rashidi, M. R. Khosravi Nikou and B. Anvaripour, Microporous Mesoporous Mater., 2015, 211, 134–141 CrossRef CAS.
- A. J. Hernandez-Maldonado and R. T. Yang, Angew. Chem., 2004, 43, 1004–1006 CrossRef CAS.
- L. Cedeño Caero, F. Jorge, A. Navarro and A. Gutiérrez-Alejandre, Catal. Today, 2006, 116, 562–568 CrossRef.
- S. Murata, K. Murata, K. Kidena and M. Nomura, Energy Fuels, 2004, 18, 116–121 CrossRef CAS.
- Y. Shiraishi, K. Tachibana, T. Hirai and I. Komasawa, Ind. Eng. Chem. Res., 2002, 41, 4362–4375 CrossRef CAS.
- H. Lu, J. Gao, Z. Jiang, Y. Yang, B. Song and C. Li, Chem. Commun., 2006, 150–152, 10.1039/b610504a.
- A. Ishihara, D. Wang, F. Dumeignil, H. Amano, E. W. Qian and T. Kabe, Appl. Catal., A, 2005, 279, 279–287 CrossRef CAS.
- S. Otsuki, T. Nonaka, N. Takashima, W. Qian, A. Ishihara, T. Imai and T. Kabe, Energy Fuels, 2000, 14, 1232–1239 CrossRef CAS.
- B. Bertleff, J. Claußnitzer, W. Korth, P. Wasserscheid, A. Jess and J. Albert, ACS Sustainable Chem. Eng., 2017, 5, 4110–4118 CrossRef CAS.
- B. Bertleff, M. S. Haider, J. Claußnitzer, W. Korth, P. Wasserscheid, A. Jess and J. Albert, Energy Fuels, 2020, 34, 8099–8109 CrossRef CAS.
- M. J. Poller, S. Bönisch, B. Bertleff, J. C. Raabe, A. Görling and J. Albert, Chem. Eng. Sci., 2022, 264, 118143 CrossRef CAS.
- M. Huber, J. Tochtermann, S. Eller, W. Korth, A. Jess and J. Albert, Energy Fuels, 2023 DOI:10.1021/acs.energyfuels.2c04136.
- B. Bertleff, J. Claußnitzer, W. Korth, P. Wasserscheid, A. Jess and J. Albert, Energy Fuels, 2018, 32, 8683–8688 CrossRef CAS.
- D. Paredes, P. Kuschk, T. S. A. Mbwette, F. Stange, R. A. Müller and H. Köser, Eng. Life Sci., 2007, 7, 13–25 CrossRef CAS.
- Z. Lei, S. Yang, L. Wang, X. Huang, X. C. Wang, Y.-Y. Li, Q. Li, Y. Zhao and R. Chen, J. Cleaner Prod., 2020, 276, 124245 CrossRef CAS.
- S. Xu, X. Wu and H. Lu, Front. Environ. Sci. Eng., 2021, 15, 133 CrossRef CAS.
- H. Daims, S. Lucker and M. Wagner, Trends Microbiol., 2016, 24, 699–712 CrossRef CAS.
- J. N. Armor, Catal. Today, 1995, 26, 147–158 CrossRef CAS.
- A. Ueda, T. Oshima and M. Haruta, Appl. Catal., B, 1997, 12, 81–93 CrossRef CAS.
- Y. Li and J. N. Armor, Appl. Catal., B, 1993, 3, 55–60 CrossRef CAS.
- G. H. Messerly and R. M. Kennedy, J. Am. Chem. Soc., 1940, 62, 2988–2991 CrossRef CAS.
- R. W. Blue and W. F. Giauque, J. Am. Chem. Soc., 1935, 57, 991–997 CrossRef CAS.
- J. P. Compton and S. D. Ward, Metrologia, 1976, 12, 101–113 CrossRef CAS.
- T. Esser, M. Huber, D. Voß and J. Albert, Chem. Eng. Res. Des., 2022, 185, 37–50 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc00648d |
| This journal is © The Royal Society of Chemistry 2023 |