
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
VITAS, a sensitive in vivo selection assay to discover enzymes producing antiviral natural products†
Aws Fahd
Alharbi
,
Hayun
Kim
,
Dhirish
Chumroo
,
Yuxuan
Ji
,
Mohammed
Hakil
and
Kourosh H.
Ebrahimi
 *
*
Institute of Pharmaceutical Science, King's College London, London, UK. E-mail: Kourosh.ebrahimi@kcl.ac.uk
First published on 11th April 2023
Abstract
To discover new broad-spectrum antiviral nucleotide analogues from natural resources or through protein engineering, we have developed a sensitive in vivo selection assay named Viral polymerase-Inhibition Toxin-Associated Selection (VITAS). We show that the assay works with enzymes from three Kingdoms of life.
There has been a rise in the emergence and re-emergence of new viruses like Zika, SARS, SARS-CoV-2, and Ebola.1 Therefore, we need to discover and develop new antiviral therapeutics. Natural products (NPs), the secondary metabolites produced by enzymes in various organisms, are an unlimited resource for finding new antiviral lead molecules.2,3 Between 1980 and 2010, more than 60% of drugs were derived from NPs.4 A prominent example of drugs originating from NPs is the antiviral nucleotide/nucleoside analogues (ANAs).5–7 Some of the earliest bioactive nucleoside analogues, e.g. pongosine, spongothymidine, and spongouridine, were derived from a marine sponge.2 These molecules were the basis for synthesising some antiviral drugs, including the anti-HIV-1 drug zidovudine.2 ANAs mimic the structure of nucleotides (the building block of RNA and DNA) and hence, inhibit or chain-terminate activity of viral polymerases. Consequently, they are among the essential antiviral drugs (> 25 FDA-approved drugs),5,7,8 and some are on the World Health Organization's List of Essential Medicines. Moreover, an analogue developed for one virus can help treat emerging viruses. For example, the ANA remdesivir was initially developed for Ebola virus and rapidly repurposed to treat infections by SARS-CoV-2.9
Recently, it has been shown that several members of the radical S-adenosylmethionine (SAM) enzymes produce a range of ANAs. Two radical-SAM enzymes were shown to produce ANA albucidin.10 We and others have shown11–13 that a new class of the radical-SAM enzymes inaccurately called prokaryotic viperin14 and thus, renamed radical-SAM dependent nucleotide dehydratases (SANDs)14 synthesise a range of new ANAs lacking 3′-hydroxyl group.
Members of the radical-SAM enzymes are widespread among all life forms11,15,16 and have a highly conserved [4Fe–4S] cluster coordinated to three cysteine residues of the CxxxCxxC motif.17,18 The [4Fe–4S]1+ cluster reductively cleaves SAM generating the 5′-deoxyadenosyl radical (5′-dA˙) intermediate. In SANDs, the 5′-dA˙ performs hydrogen atom transfer (HAT) from a nucleoside triphosphate (NTP) with an exquisite degree of selectivity, producing 3′-deoxy-3′,4′-didehydro-NTPs like ddhUTP and ddhCTP (Fig. 1a). The ddhCTP analogue has antiviral activity against a large number of viruses and modulates the immune response.19,20 Some fungal and microbial SANDs are shown to perform a similar chemical reaction synthesising ddhUTP, ddhCTP, and ddhGTP.12,13 Therefore, SANDs or similar enzymes are a resource for discovering antiviral NPs to help develop new therapeutics.
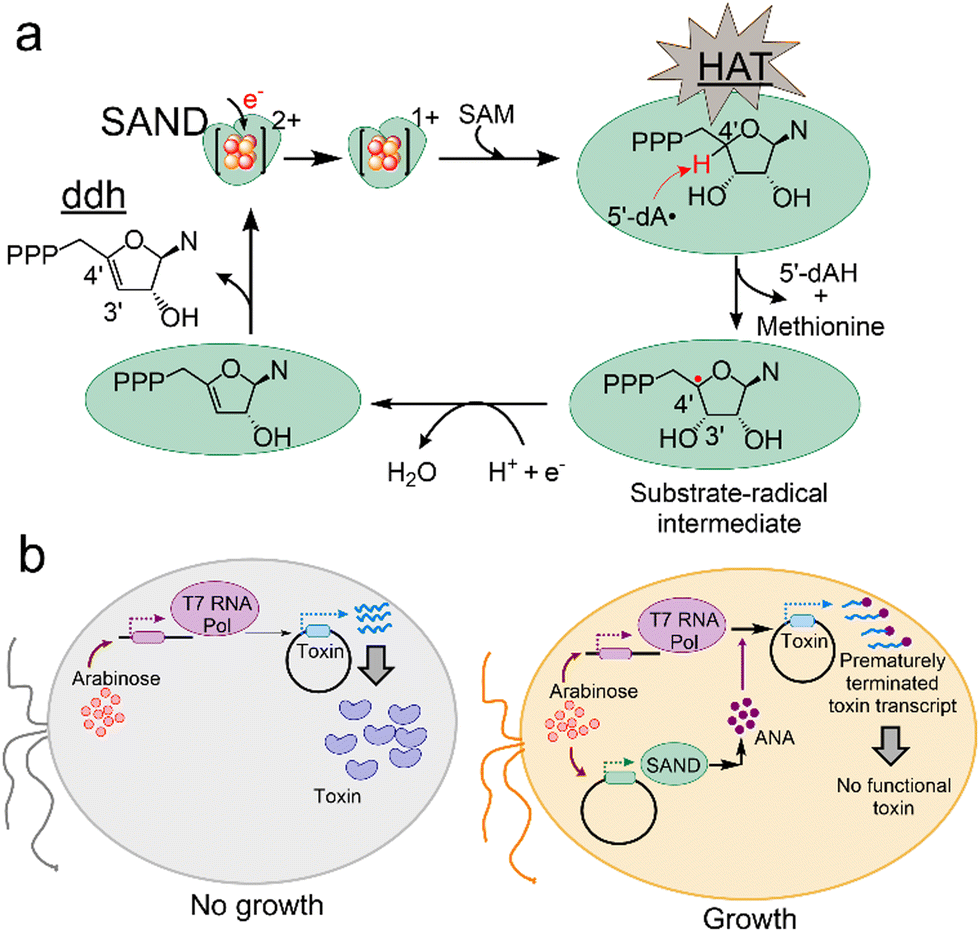 | ||
| Fig. 1 Principles of the VITAS assay for discovering antiviral enzymes and NPs. (a) Members of SANDs catalyse the transformation of a nucleotide to a ddh nucleotide analogue via a HAT step. This process requires one-electron reduction of the [4Fe–4S]2+ cluster and subsequent reductive cleavage of SAM. N, nucleobase. 5′-dAH: 5′-deoxyadenosine. (b) The VITAS assay. In an engineered E. coli, the expression of viral T7 RNA polymerase (Pol) is induced by adding the first inducer, arabinose. T7 polymerase mediates the expression of a toxin protein under the control of the leaky T7 promotor (left). Consequently, cell growth is blocked. When the cells are transformed with a plasmid expressing a SAND (right), upon addition of arabinose, SAND activity leads to the synthesis of an ANA in E. coli, prematurely terminating the activity of T7 polymerase and blocking the synthesis of a functional toxin. As a result, cell growth is rescued. | ||
To discover new enzymes or engineer existing SANDs and expand their chemical space for the synthesis of new ANAs, we perceived a novel assay named VITAS (Viral polymerase-Inhibition Toxin-Associated Selection) (Fig. 1b). The VITAS assay is based on the hypothesis that an ANA is formed due to the expression and activity of an enzyme in E. coli. The ANA inhibits viral T7 RNA polymerase (Pol)-mediated expression of a toxin protein allowing cell growth (Fig. 1b). The VITAS assay fundamentally differs from the commonly used live/dead assays in drug discovery. Traditional assays rely on chemical or biological labelling of cells during/after treatment with known purified lead molecules synthesised either chemically or using a purified enzyme. Moreover, purifying oxygen-sensitive radical-SAM enzymes is not straightforward, limiting the use of in vitro assays. These commonly used assays cannot easily be adopted for protein engineering to rapidly screen the activity of enzyme variants in a large library. In sharp contrast, the VITAS assay eliminates the need to purify enzymes and the NPs. Therefore, it enables the screening of enzymes’ activity, facilitating the mining of the repository of natural enzymes and protein engineering to discover new ANAs. The VITAS assay is sensitive to the SANDs' activity, unlike the previously reported fluorescence-based assay showing similar activity for human and microbial SANDs.12
To test our hypothesis and create the assay, we used the E. coli BL21-AI strain, in which the expression of T7 RNA Pol is under the tightly-regulated arabinose-inducible PBAD promotor.21 We decided to test the toxin LdrD protein from the Type-I toxin/antitoxin LdrD/RdiD system because its expression rapidly inhibits E. coli cell growth.22 The LdrD gene was cloned into pET28a (ESI† Methods) under the control of the leaky T7 promotor.23 In the pET expression system, the lacI repressor blocks the expression of a target gene in the absence of inducer IPTG (Isopropyl β-d-1-thiogalactopyranoside). However, this repression is not 100%, and the target gene has a background expression (i.e. the promoter is leaky). Upon addition of IPTG, the repressor is inactivated, and T7 polymerase can bind to its promotor, inducing the target gene expression. To confirm that the LdrD expression effectively blocks E. coli growth, we transformed E. coli BL21-AI cells with the LdrD expression plasmid or, as a control, a plasmid expressing the thermostable SAND from the fungal Thielavia terrestris (TtSAND). As described before, TtSAND was cloned in pBAD/His C plasmid,24 and its expression is under the tightly-regulated PBAD promotor.21 After transformation, cells were spread on LB-Agar plates containing arabinose and IPTG. Then, the plates were incubated overnight at 37 °C (ESI† Methods). Visible white colonies were observed for cells expressing TtSAND but not for the LdrD-expressing cells (Fig. S1, ESI†), confirming that the LdrD overexpression effectively blocks the growth.
Subsequently, we tested if the overexpression of a known SAND can interfere with the activity of T7 RNA Pol and, thus, the expression and toxicity of LdrD. We first tested the fungal TtSAND because it can generate ddhCTP and ddhUTP.11E. coli BL21-AI cells were transformed with plasmids encoding TtSAND, LdrD, or both TtSAND and LdrD (SI Methods) (Fig. 2a). We developed the VITAS assay using a simple 3-step procedure (Fig. 2b). After transformation, the cells were spread on LB-Agar plates containing 0.02% arabinose to induce the expression of TtSAND and T7 RNA Pol (ESI† Methods). Plates were incubated overnight at 37 °C. The next day, single colonies were picked from each plate and inoculated in 2 mL LB media containing 1 mM IPTG. Cells were incubated in a shaker at 200 rpm and 37 °C. After 6 hours, the growth of E. coli cells was recorded at 600 nm (Fig. 2c). The growth was rescued when cells were co-transformed with TtSAND and LdrD plasmids (Fig. 2c). This finding confirms that the TtSAND expression effectively blocked T7 RNA Pol-mediated expression of toxin LdrD. Therefore, the ddh nucleotide analogue produced by TtSAND acts as an ANA or is further modified by E. coli enzymes generating an antiviral molecule inhibiting the T7 RNA Pol activity.
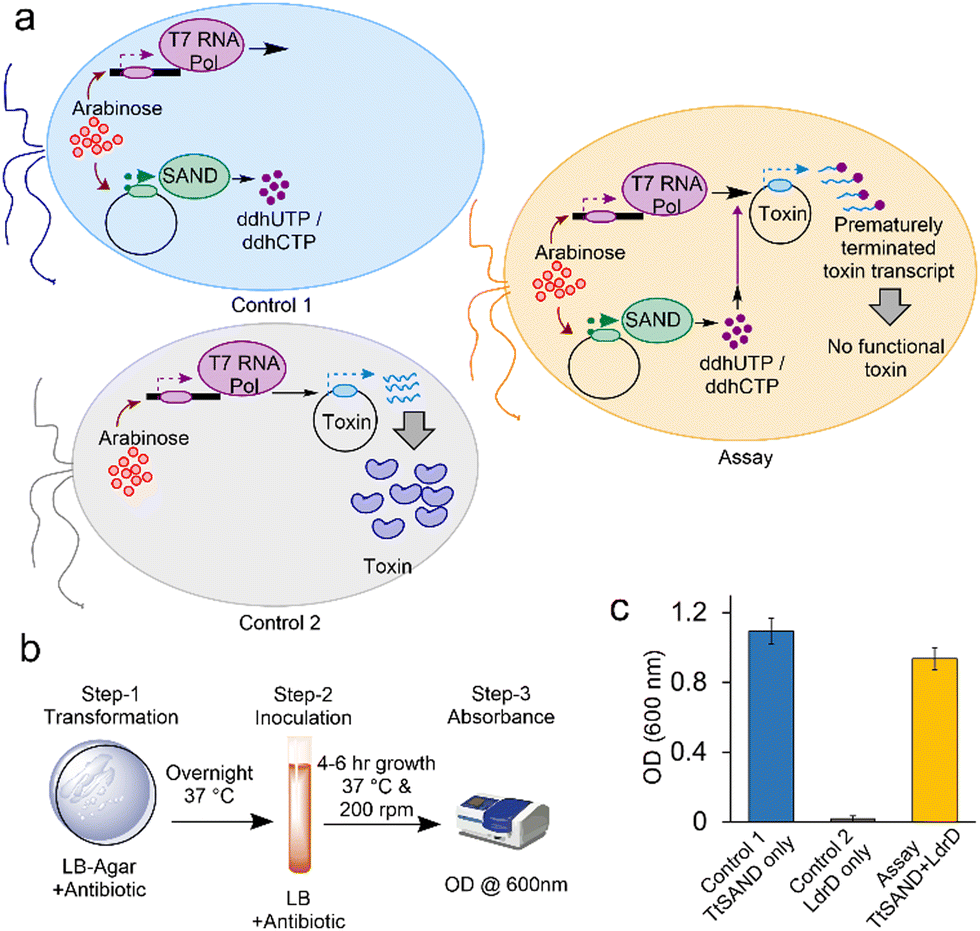 | ||
| Fig. 2 Fungal TtSAND inhibits T7 RNA Pol-mediated expression of toxin LdrD. (a) A schematic presentation of the VITAS assay and two controls. (b) Three-step assay developed to use VITAS. (c) The TtSAND expression, which produces ddhUTP/ddhCTP nucleotide analogues, effectively blocks the synthesis of active LdrD by T7 RNA Pol and allows the growth of E. coli cells. The assay was performed using 0.02% arabinose in LB-Agar plates (step-1) and 1 mM IPTG in growth media (step-2). Data are the average of three independent replicates ± standard deviations. | ||
Next, we aimed to test if we would obtain similar results with human SAND (hSAND) producing ANA ddhCTP.25,26 We varied arabinose or IPTG concentrations (Fig. S2, ESI†) to modulate the hSAND and T7 RNA Pol expressions and activities or the LdrD expression and toxicity, respectively. When arabinose was not added (0.0% arabinose) to the LB-Agar plates (Fig. 3), the growth was the same within experimental error for the cells transformed with hSAND, LdrD or both hSAND and LdrD plasmids (Fig. 3). In the absence of arabinose, the growth was independent of the IPTG amount added to the LB medium (Fig. S2, ESI†). Therefore, the addition of arabinose is required to induce the T7 RNA Pol and the toxin LdrD expressions. The amount of arabinose could modulate the T7 RNA Pol activity and the LdrD expression and toxicity in the absence of IPTG (Fig. 3). The more the concentration of arabinose, the higher T7 RNA Pol expression and activity, thus, the more the LdrD expression and toxicity (Fig. 3). These observations are consistent with the fact that the T7 promotor is leaky.23 As the arabinose concentration increased from 0.002% to 0.02%, the growth of hSAND-only expressing cells decreased (Fig. 3). A toxicity test showed that hSAND expression reduced E. coli growth (Fig. S3, ESI†). The hSAND expression in E. coli affects cell morphology.27 These observations suggest that the hSAND overexpression is toxic to E. coli but significantly less than that of the LdrD overexpression (Fig. 3). At 0.02% arabinose with no IPTG added, the growth of LdrD-only expressing cells was almost entirely blocked. However, cells transformed with both hSAND and LdrD could grow (Fig. 3). We conclude that the nucleotide analogue ddhCTP produced by hSAND either directly or indirectly inhibits the activity of T7 RNA Pol and the synthesis of the functional toxin LdrD protein. This conclusion is consistent with earlier observations that hSAND expression in human cells suppresses T7 promotor-dependent RNA synthesis.28
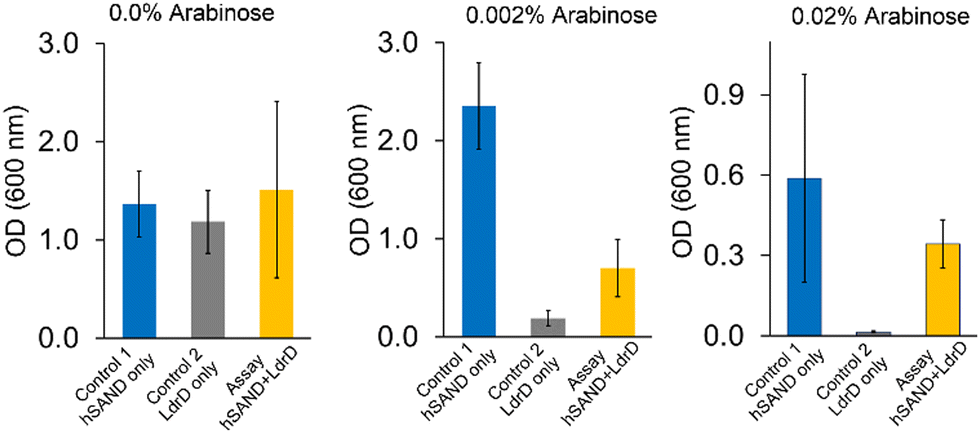 | ||
| Fig. 3 hSAND inhibits T7 RNA Pol activity in the VITAS assay. The addition of arabinose is enough to induce the expression and toxicity of LdrD due to the leaky T7 promotor. Three different concentrations of arabinose in the LB-Agar plates were tested in the absence of IPTG. Data are the average of three independent replicates ± standard deviations. | ||
After establishing the VITAS assay using human and fungal SANDs, we used the assay to identify new antiviral enzymes. As an example, we selected a putative bacterial SAND from Pseudodesulfovibrio piezophilus (PpSAND), which we predicted to be a homologue of TtSAND.11 The AlphaFold-predicted structure of the enzyme (Fig. 4a) shows the CxxxCxxC motif forming the coordination environment required for the [4Fe–4S] cluster binding and the GGE motif of the SAM binding pocket (Fig. 4a). The expression of PpSAND by itself in E. coli did not cause toxicity (Fig. S4, ESI†). Next, we used the VITAS assay to screen the antiviral activity of PpSAND. We found that transforming cells with PpSAND and LdrD plasmids improved the growth of E. coli cells compared to the LdrD-only expressing cells (Fig. 4b). Therefore, PpSAND expression partially inhibited the T7 RNA Pol activity, reducing the LdrD expression. The ability of PpSAND to inhibit the T7 RNA Pol-mediated expression of LdrD and improve growth was affected by the concentration of IPTG (Fig. S5, ESI†), like what we observed for hSAND (Fig. S2, ESI†). At 1 mM IPTG, PpSAND was not able to inhibit the activity of T7 RNA Pol and the expression of toxin LdrD protein (Fig. 4b), unlike TtSAND (Fig. 2c). Additionally, the ability of PpSAND to rescue the E. coli growth was less than that of TtSAND. The exact reason for this observation is unclear. It could be because of: (i) the lower efficacy of the ANA produced by this enzyme, or (ii) its inefficient catalytic activity. The activity of PpSAND might be less due to the low soluble fraction of this protein inside E. coli cells. Although we could purify PpSAND (Fig. S6, ESI†) like TtSAND as we reported previously,24 the protein solubility and purification could have been affected by lysis buffer or sonication. Alternatively, the low activity of PpSAND could be due to inefficient electron transfer from a yet-to-be-identified partner or [4Fe–4S] cluster transfer from the E. coli iron-sulfur biogenesis machinery. We used UV-visible spectroscopy to test the presence of [FeS] cluster in PpSAND. As a control, we measured the absorbance spectrum of TtSAND, which we previously confirmed to contain a [4Fe–4S] cluster.11,24 The PpSAND UV-visible spectrum showed an absorbance peak around 405–410 nm, like TtSAND (Fig. 4c). These findings corroborate that PpSAND contains a [4Fe–4S] cluster. The amino acid blast analysis shows that PpSAND has several uncharacterised homologues in various organisms (Fig. S7, ESI†). Further studies using liquid chromatography-mass spectrometry and NMR spectroscopy are required to identify and characterise the ANA produced by PpSAND.
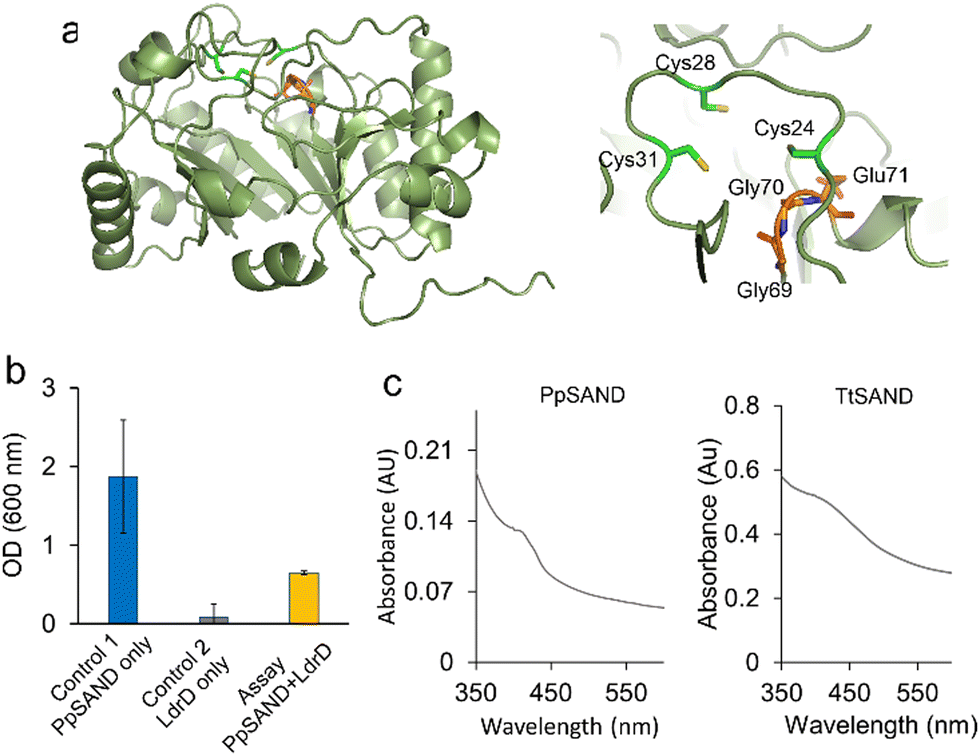 | ||
| Fig. 4 Discovery of a new SAND using the VITAS assay. (a) The AlphaFold-predicted structure of PpSAND. The predicted structure has the highly conserved CxxxCxxC shown to bind to the [4Fe–4S] cluster and the GGE motif of the SAM binding pocket. (b) The VITAS assay shows that a putative SAND enzyme from the bacterium Pseudodesulfovibrio piezophilus (PpSAND) produces an antiviral lead NP inhibiting activity of viral T7 RNA Pol. Data are the average of measurements using three biologically independent samples ± standard deviations. (c) PpSAND contains a [4Fe–4S] cluster like TtSAND. | ||
In summary, we demonstrated the capabilities of the VITAS assay and its applicability to investigate fungi, humans, and bacteria enzymes producing ANAs. We tested only a single toxin protein LdrD. Likely, other types of toxins from type-I, -II, and -III can be used. The assay is sensitive to the activity of enzymes. Specifically, by varying the arabinose or IPTG concentration, we modulated the T7 RNA Pol expression and activity, thus, the LdrD expression and toxicity. Under varying arabinose or IPTG concentrations, each SAND showed a different response. It inhibited the T7 RNA Pol activity and the LdrD toxicity at a different level. Variation in the activity of enzymes could be due to multiple factors: (i) the difference in the efficacy of ANAs in blocking the viral T7 RNA Pol activity, (ii) the difference in electron transfer barrier29 or [FeS] cluster transfer from the E. coli iron-sulfur biogenesis machinery leading to different catalytic efficiency, and/or (iii) variation in the soluble fraction of enzymes inside E. coli cells. Further work is required to explore each factor's contribution to in vivo activity, couple the assay to an LC-MS instrument, and modify it for high-throughput screening (HTS). We predict that the VITAS assay will facilitate the screening of enzyme variants to discover or engineer SANDs and other novel enzymes producing ANAs. The discovery of new enzymes is expected to help identify novel ANA NPs and expand the chemical space accessible for the enzymatic or chemoenzymatic synthesis of new antivirals.
KHE, Conceptualization, funding acquisition, and supervision; AFA, HK, DC, YJ, and MA, Data curation; KHE, writing the original draft with contribution from all co-authors; KHE, review & editing. KHE is grateful to Prof. Analise Pastore for generously dedicating her anaerobic glovebox to his lab. KHE thanks Dr Yvain Nicolet (ibs, France) for kindly providing pET28a vector and E. coli BL21-AI cells. This project was supported by King's College London student funds, the Royal Society International Exchange Grant (IES\R1\221117), and the Royal Society Research Grant (RGS\R1\231135) to KHE. All authors acknowledge the support from the European Cooperation in Science and Technology (COST) Action CA21115.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. J. Carlson, G. F. Albery, C. Merow, C. H. Trisos, C. M. Zipfel, E. A. Eskew, K. J. Olival, N. Ross and S. Bansal, Nature, 2022, 607, 555–562 CrossRef CAS PubMed.
- V. Gogineni, R. F. Schinazi and M. T. Hamann, Chem. Rev., 2015, 115, 9655–9706 CrossRef CAS PubMed.
- J. Li, Y. Wang, X. Hao, S. Li, J. Jia, Y. Guan, Z. Peng, H. Bi, C. Xiao, S. Cen and M. Gan, Molecules, 2019, 24, 2821 CrossRef CAS PubMed.
- F. I. Saldívar-González, V. D. Aldas-Bulos, J. L. Medina-Franco and F. Plisson, Chem. Sci., 2022, 13, 1526–1546 RSC.
- L. P. Jordheim, D. Durantel, F. Zoulim and C. Dumontet, Nat. Rev. Drug Discovery, 2013, 12, 447–464 CrossRef CAS PubMed.
- S. Mahmoud, S. Hasabelnaby, S. Hammad and T. Sakr, J. Adv. Pharm. Res., 2018, 2, 73–88 CrossRef.
- U. Pradere, E. C. Garnier-Amblard, S. J. Coats, F. Amblard and R. F. Schinazi, Chem. Rev., 2014, 114, 9154–9218 CrossRef CAS PubMed.
- R. J. Geraghty, M. T. Aliota and L. F. Bonnac, Viruses, 2021, 13, 667 CrossRef CAS PubMed.
- J. H. Beigel, K. M. Tomashek, L. E. Dodd, A. K. Mehta, B. S. Zingman, A. C. Kalil, E. Hohmann, H. Y. Chu, A. Luetkemeyer and S. Kline, N. Engl. J. Med., 2020, 383, 1813–1836 CrossRef CAS PubMed.
- P.-H. Fan, Y. Geng, A. J. Romo, A. Zhong, J. Zhang, Y.-C. Yeh, Y.-H. Lee and H.-W. Liu, Angew. Chem., Int. Ed., 2022, 134, e202210362 Search PubMed.
- K. H. Ebrahimi, J. Rowbotham, J. McCullagh and W. S. James, ChemBioChem, 2020, 21, 1605–1612 CrossRef PubMed.
- A. Bernheim, A. Millman, G. Ofir, G. Meitav, C. Avraham, H. Shomar, M. M. Rosenberg, N. Tal, S. Melamed, G. Amitai and R. Sorek, Nature, 2021, 589, 120–124 CrossRef CAS PubMed.
- J. C. Lachowicz, A. S. Gizzi, S. C. Almo and T. L. Grove, Biochemistry, 2021, 60, 2116–2129 CrossRef CAS PubMed.
- Y. Ji, W. Li, A. Da, H. Stark, P.-L. Hagedoorn, S. Ciofi-Baffoni, S. A. Cowley, R. O. Louro, S. Todorovic and M. A. Mroginski, Front. Mol. Biosci., 2022, 9, 1032220 CrossRef CAS PubMed.
- K. H. Ebrahimi, D. Howie, J. Rowbotham, J. McCullagh, F. Armstrong and W. S. James, FEBS Lett., 2020, 594, 1624–1630 CrossRef CAS PubMed.
- M. K. Fenwick, Y. Li, P. Cresswell, Y. Modis and S. E. Ealick, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6806–6811 CrossRef CAS PubMed.
- J. B. Broderick, B. R. Duffus, K. S. Duschene and E. M. Shepard, Chem. Rev., 2014, 114, 4229–4317 CrossRef CAS PubMed.
- K. H. Ebrahimi, Metallomics, 2018, 10, 539–552 CrossRef PubMed.
- K. H. Ebrahimi, S. Diofi, P. L. Hagedoorn, Y. Nicolet, N. E. Le Brun, W. R. Hagen and F. A. Armstrong, Nat. Chem., 2022, 14, 253–266 CrossRef PubMed.
- S. Ghosh and E. N. G. Marsh, J. Biol. Chem., 2020, 295, 11513–11528 CrossRef CAS PubMed.
- L.-M. Guzman, D. Belin, M. J. Carson and J. O. N. Beckwith, J. Bacteriol., 1995, 177, 4121–4130 CrossRef CAS PubMed.
- M. Kawano, T. Oshima, H. Kasai and H. Mori, Mol. Microbiol., 2002, 45, 333–349 CrossRef CAS PubMed.
- G. J. Gopal and A. Kumar, Protein J., 2013, 32, 419–425 CrossRef CAS PubMed.
- K. H. Ebrahimi, S. B. Carr, J. McCullagh, J. Wickens, N. H. Rees, J. Cantley and F. Armstrong, FEBS Lett., 2017, 591, 2394–2405 CrossRef PubMed.
- A. S. Gizzi, T. L. Grove, J. J. Arnold, J. Jose, R. K. Jangra, S. J. Garforth, Q. Du, S. M. Cahill, N. G. Dulyaninova, J. D. Love, K. Chandran, A. R. Brenick, C. E. Cameron and S. C. Almo, Nature, 2018, 558, 610–614 CrossRef CAS PubMed.
- K. H. Ebrahimi, J. Vowles, C. Browne, J. McCullagh and W. S. James, FEBS Lett., 2020, 594, 1631–1644 CrossRef PubMed.
- M. T. Nelp, A. P. Young, B. M. Stepanski and V. Bandarian, Biochemistry, 2017, 56, 3874–3876 CrossRef CAS PubMed.
- A. Dukhovny, A. Shlomai and E. H. Sklan, Sci. Rep., 2018, 8, 8100 CrossRef PubMed.
- F. D’Angelo, E. Fernández-Fueyo, P. S. Garcia, H. Shomar, M. Pelosse, R. R. Manuel, F. Büke, S. Liu, N. van den Broek and N. Duraffourg, eLife, 2022, 11, e70936 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc00638g |
| This journal is © The Royal Society of Chemistry 2023 |