
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-enzymatic protein templates amide bond formation and provides catalytic turnover†
Nicolas
Brauckhoff
ab,
Laura
Fang
ab,
Anissa
Haim
cd and
Tom N.
Grossmann
 *abcd
*abcd
aChemical Genomics Centre of the Max Planck Society, Dortmund, 44227, Germany
bDepartment of Chemistry and Chemical Biology, Technical University Dortmund, Dortmund, 44227, Germany
cDepartment of Chemistry and Pharmaceutical Sciences, Vrije Universiteit Amsterdam, Amsterdam, 1081 HZ, The Netherlands
dAmsterdam Institute of Molecular and Life Sciences (AIMMS), Vrije Universiteit Amsterdam, Amsterdam, 1081 HZ, The Netherlands. E-mail: t.n.grossmann@vu.nl
First published on 6th April 2023
Abstract
The spatial alignment of functional groups is a central aspect of most catalytic processes. Protein scaffolds with their exceptional molecular recognition properties have evolved into powerful biological catalysts. However, the rational design of artificial enzymes starting from non-catalytic protein domains proved challenging. Herein, we report the use of a non-enzymatic protein as template for amide bond formation. Starting from a protein adaptor domain capable of simultaneously binding to two peptide ligands, we designed a catalytic transfer reaction based on the native chemical ligation. This system was used for the selective labelling of a target protein validating its high chemoselectivity and potential as a novel tool for the selective covalent modification of proteins.
Accelerating chemical reactions via the alignment of functional groups is a hallmark of catalytic processes.1 A central characteristic of a catalyst is its potential to facilitate repetitive reaction cycles generating multiple turnovers.2–4 The catalytic efficiency depends on a multitude of factors including the rate of the chemical reaction and the affinity of substrates as well as reaction products.1 While nature evolved a multitude of very efficient enzyme catalysts, the design of catalytic proteins from non-enzymatic domains represents a major challenge.5–7 Due to the high abundance of amides in biological systems and their importance to the origin of life, the protein-mediated formation of amide bonds gained attention.8–10 Naturally occurring enzymes such as ligases, transpeptidases and proteases have been utilised for amidation reactions, however, limited by their inherent substrate spectrum.11–13 As an alternative approach, peptide-based coiled coils served as basis for the design of templated amide bond formations.14–16 In addition, the tetrameric protein streptavidin was used to template amide formations between desthiobiotin modified substrates.17 In these examples, catalytic activities were low due to slow exchange between products and substrates.
We have previously designed a templated peptide ligation employing a thiol-selective bioconjugation reaction.18 In this setup, the two peptide substrates bind simultaneously to the KIX domain of the human CREB binding protein, which thereby served as template (T, Fig. 1A). The two simultaneously binding peptides originated from the mixed-lineage leukemia protein (ligand 1) and from the cAMP response element-binding protein (CREB, ligand 2). In the ternary complex, the N-termini of both peptides are aligned in spatial proximity,19,20 thereby triggering a reaction between introduced functional groups to furnish a thioether bridge.18 The resulting ligation product exhibited increased binding affinity for the protein template, which prevented catalytic turnover.
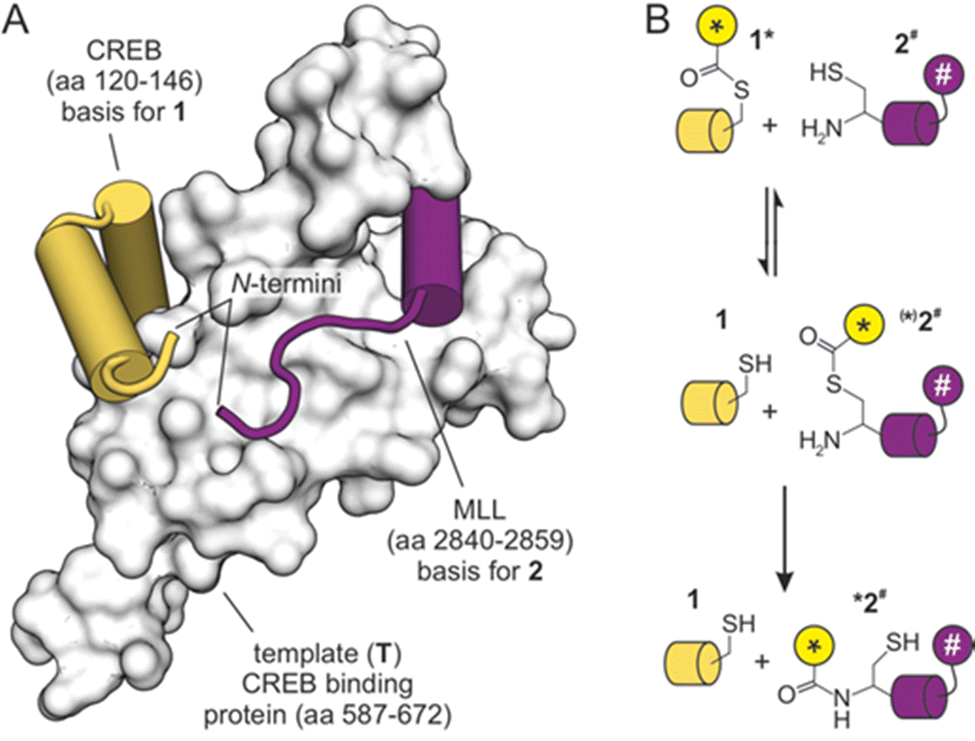 | ||
| Fig. 1 (A) NMR-structure of the KIX domain bound to peptides derived from CREB and MLL (PDB ID 2lxt). N-termini of both peptides are indicated. (B) Principle of template-mediated transfer reaction based on the native chemical ligation (* transferrable fluorescent group, # fluorescence quencher). | ||
Herein, we aim for the reduction of product inhibition in a protein templated amide bond formation, thereby enabling dissociation of the reaction products and subsequent binding of new reactants. Notably, most templated reactions accelerate a chemical transformation that otherwise would only proceed very slowly (background reaction).14–16 The background reaction, however, accelerates with increasing reactant concentrations, which renders templated reactions most useful at relatively high dilutions. For that reason, they usually find application where reactants and template are present at low concentrations, such as the detection or labelling of bio-macromolecules. In this study, we report the use of a transfer reaction (instead of the ligation) to omit the generation of products with increased template affinity. This enables catalytic turnover and the selective labelling of a target protein.
We selected the so-called native chemical ligation (NCL), a reaction that was originally used for linking two peptide fragments.21–23 To enable a transfer reaction, peptides were modified in such a way, that a transferrable group (*) was linked via a thioester to a thiol side chain in ligand 1 (Fig. 1B). Peptide ligand 2 was equipped with an N-terminal cysteine capable of undergoing first a reversible thiol exchange that initiates the transfer reaction which is concluded by an irreversible S→N-acyl shift (Fig. 1B).24,25 To facilitate a real-time readout of the reaction, a fluorophore was selected as transferrable group (*), and accepting ligand 2 was equipped with a fluorescence quencher (#). The transfer of the fluorophore-containing transferrable group to quencher-containing peptide 2 brings both chromophores in close proximity and would result in reduced fluorescence intensity.
Initially, the peptide ligands were adjusted to the planned reaction setup. The original sequence of ligand 1 contained a serine (S) close to the anticipated thioester site and two additional lysine residues (K, ESI† Fig. S2). To reduce the nucleophilic character of this peptide and exclude an intramolecular attack of the thioester moiety, these three amino acids were substituted by alanine (A). Fluorescence polarization binding assays with fluorescein isothiocyanate (FITC) labelled peptides showed that these variations did not affect affinity for template T (Kd = 0.16 ± 0.01 μM, ESI† Fig. S2). Peptide 1 was equipped with an N-terminally acetylated cysteine (C) that was subsequently loaded with a transferrable group derived from FITC, to provide the final thioester containing ligand 1* (Fig. 2A). In brief, these modifications were introduced on solid-support after peptide sequence assembly26 by adding a 4-monomethoxy-trityl side chain-protected cysteine. The cysteine side chain was then selectively deprotected and coupled with FITC-modified 5-aminovaleric acid which was obtained following a previously reported procedure.27 The sequence of peptide 2 was slightly truncated compared to the original ligation reaction,18 resulting in a 17-mer peptide (Kd = 0.50 ± 0.05 μM, ESI† Fig. S3). To obtain the final reactant 2# (Fig. 2A), this sequence was equipped with an N-terminal cysteine and a C-terminal 4-(4-dimethylaminophenylazo)benzoyl (Dabcyl) modified lysine acting as the fluorescence quencher.
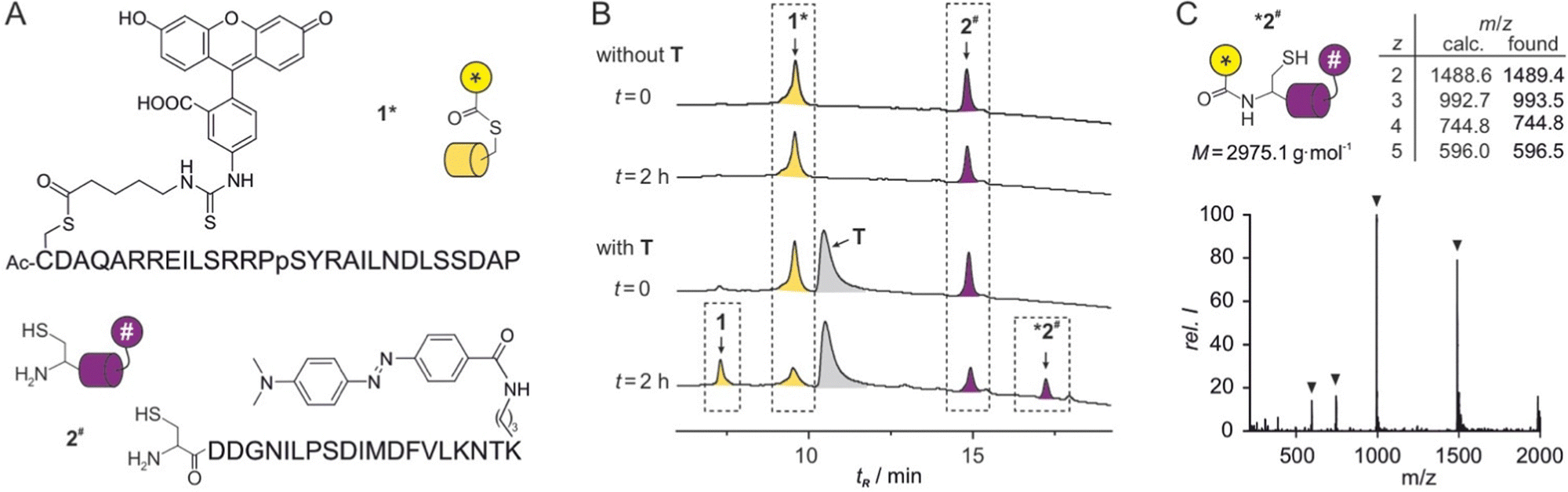 | ||
| Fig. 2 (A) Sequences with chemical structures of modifications of peptides 1* and 2#. (B) HPLC chromatograms of reactions between 1* and 2# in the absence (top) and the presence (bottom) of protein T (conditions: T = 30 °C, phosphate buffer, pH 7.4, c = 2.5 μM 1*, 5 μM 2#, 2.5 μM T, for details see ESI†). (C) MS spectrum of *2# including calculated and found m/z values. | ||
To analyse the transfer reaction, initially HPLC/MS was used as readout choosing relatively low peptide concentrations to limit a potential background reaction. In the absence of T, co-incubation of peptide 1* (c = 2.5 μM) and 2# (c = 5 μM) did not result in the formation of a reaction product (t = 2 h, Fig. 2B, top). In the presence of stochiometric amounts of T (c = 2.5 μM), two new signals appeared while the signals for 1* and 2# showed reduced intensity (t = 2 h, Fig. 2B, bottom). HPLC-coupled ESI-MS analysis associated the expected two reaction products 1 and *2# with these new signals (Fig. 2C and ESI† Fig. S6). Peak areas indicated a yield of ca. 50% after these 2 h reaction time.
Having verified the functionality of the reaction system, we next pursued a fluorescence-based reaction readout. Given the absence of detectable background reaction in the previous setup after two hours (Fig. 2B), we decided to apply higher peptide concentrations to support catalytic turnover and increase fluorescence signal intensity. The reduction in fluorescence intensity upon formation of product *2# (Fig. 3A) was monitored using a microtiter plate reader. Initially, 1* (c = 5 μM) and 2# (c = 10 μM) were reacted in the absence of T revealing the expected unchanged fluorescence intensity over time (grey, Fig. 3B). In the presence of T (c = 1.25 μM, 0.25 eq.), we observed a time-dependent intensity reduction. Notably, 0.25 eq. of T resulted in a strong signal reduction after 5 h (black, Fig. 3B) indicating the turnover of more than one substrate pair. Here, a single turnover number (TON)28 would result in only 25% yield. To investigate the catalytic activity in more detail, the reaction was performed in the presence of different concentrations of T and processed to allow the calculation of reaction yields (for details see ESI†). Importantly, in presence of substochiometric amounts of T (0.25, 0.1 and 0.05 eq.), we observed catalytic activity with TON values ranging from 3.6 to 5.0 (Fig. 3C).
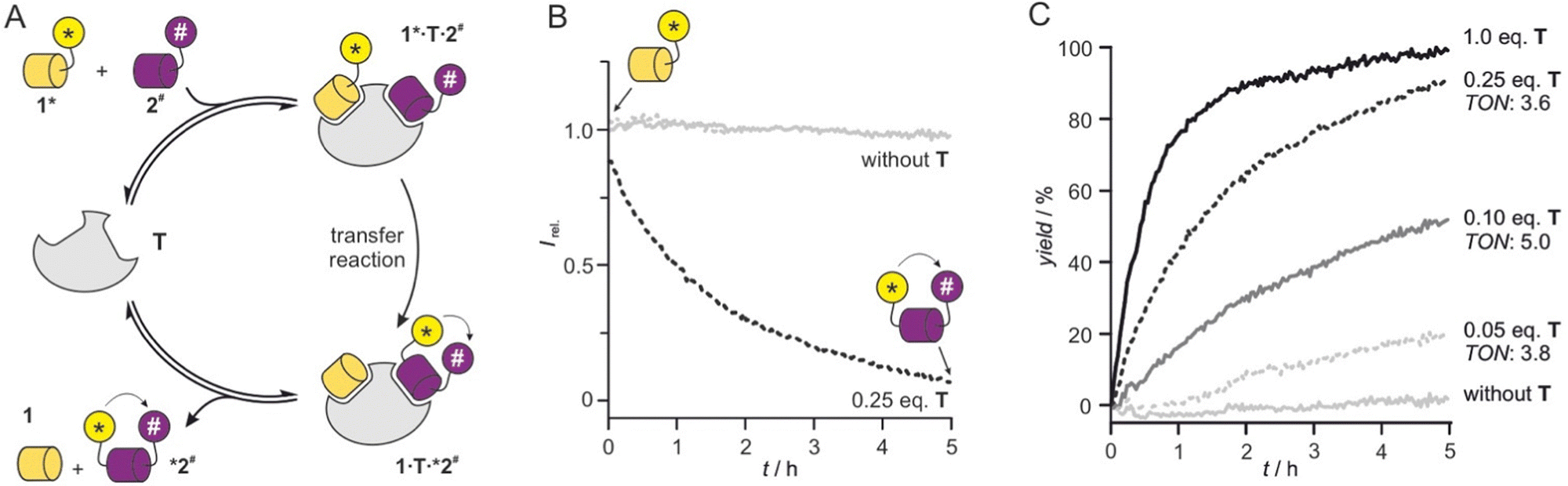 | ||
| Fig. 3 (A) Scheme of catalytic cycle starting from 1* and 2# and providing reaction products 1 and *2#. (B) Fluorescent readout of transfer reaction (conditions: T = 30 °C, phosphate buffer, pH 7.4, c = 5 μM 1*, 10 μM 2#, 1.25 μM T (0.25 eq.), for details see ESI†). (C) Reaction time course determined based on fluorescence intensity changes for different equivalents of T (conditions: T = 30 °C, phosphate buffer, pH 7.4, c = 5 μM 1*, 10 μM 2#, for details see ESI†). Turnover numbers (TON) are provided (for details ESI† Fig. S7). | ||
Usually, the reduction of catalyst equivalents increases TON values. However, for the lowest concentration of T (c = 0.25 μM, 0.01 eq., Fig. 3C), we did not observe the highest TON. This points towards insufficient complex formation and/or ligand exchange under these conditions, which prompted us to test higher peptide concentrations (c = 50 μM 1* and 75 μM 2#). And indeed, under these conditions TON values increased to 8.8 for 0.05 eq. T (c = 2.5 μM) after 5 h (ESI† Fig. S8). In the presence of 0.01 eq. T (c = 0.5 μM), a TON value of 16 was obtained after 5 h. Notably, under these conditions a considerable background reaction was observed (ca. 50% yield in the absence of T, ESI† Fig. S8) owing to the overall high substrate concentration.
Knowing about the high chemo-selectivity of the NCL reaction and its preference for N-terminal free cysteine residues, we considered the use of 1* and T for protein labelling. To test the feasibility of this approach, we chose firefly luciferase from Photinus pyralis as the protein of interest (POI) and equipped its N-terminus with peptide sequence 2, including the terminal free cysteine resulting in protein 2-POI. In brief, a GST tag was cloned N-terminally to 2-POI linking both constructs via a TEV protease recognition motif.29 The TEV site was modified to provide the required N-terminal free cysteine in 2-POI after protease cleavage (for details see ESI†).
For labelling reactions, we applied low reactant concentrations to reduce the risk of undesired side reactions with alternative nucleophilic protein side chains. Target protein 2-POI (c = 1 μM) was then co-incubated with 1* (c = 1 μM) either in the presence or absence of T (Fig. 4A). To follow the time course of the transfer reaction, aliquots were subjected to SDS loading buffer and then analysed using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). To visualise protein labelling, the resulting gel was imaged using a fluorescence reader (Fig. 4B, top) and subsequently analysed via Coomassie staining (Fig. 4B, bottom). The band intensity in the fluorescence image (Fig. 4B, top) is indicative of labelled protein *2-POI clearly showing a time-dependent increase in the presence of T. In the absence of T, we did not observe labelling under these conditions (t = 120 min). The Coomassie-stained gel shows a total protein content irresectable of the labelling status. Here, similar band intensities in all lanes were observed (Fig. 4B, bottom), indicating efficient protein labelling. To investigate if labelling occurs at the N-terminal cysteine, a C1A variant of 2-POI was expressed and tested showing indeed the expected absence of a fluorescence signal (ESI† Fig. S11). Also, enzymatic luciferase activity of 2-POI before and after the labelling was assessed revealing unchanged activity (ESI† Fig. S12). Taken together, this confirms the successful formation of the covalently labelled protein *2-POI.
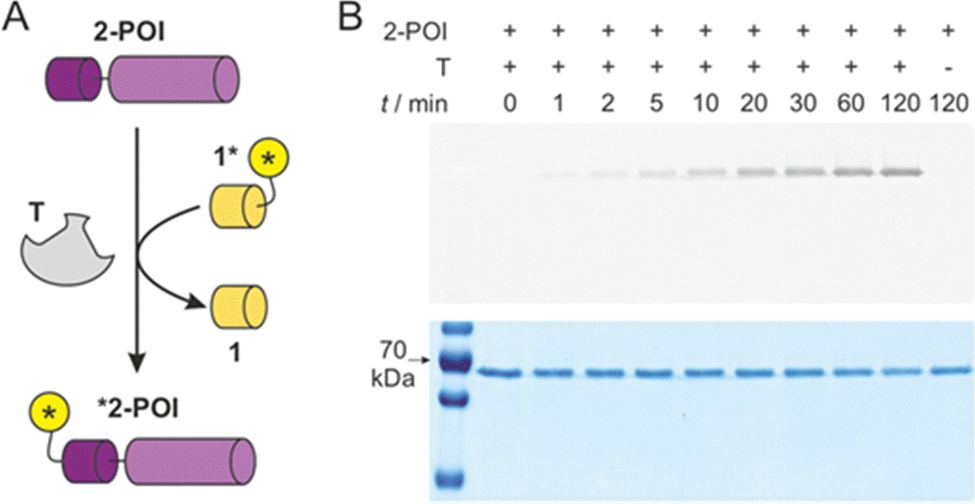 | ||
| Fig. 4 (A) Scheme of T-mediated labelling of protein 2-POI (for protein sequence see ESI† Fig. S1). (B) SDS-PAGE analysis of time-dependence of labelling reaction with 1*, 2-POI and T (each c = 1 μM, buffer: 20 mM sodium phosphate, pH 7.4, 500 μM TCEP, T = 30 °C). Top: Fluorescence imaging of gel indicating labelled protein *2-POI, Bottom: Coomassie-stained gel indicative of total protein content 2-POI and *2-POI (for full gels see ESI† Fig. S10). | ||
In conclusion, we report the use of a protein adaptor domain, capable of simultaneously binding to two peptide ligands, as template for amide formation. In a related previously reported system, the protein domain was used to template a ligation reaction providing a product with high template affinity.18 This resulted in severe product inhibition and hence did not allow catalytic turnover. Herein, we report the design of a pair of peptide ligands that can undergo a transfer reaction following a mechanism analogous to the native chemical ligation. The transferrable group harbours a fluorophore which facilitated (i) a real-time readout via transfer to a fluorescence quencher-bearing peptide and (ii) the labelling of a protein equipped with a suitable N-terminal peptide tag. The highest observed TON value was 16 (0.01 eq. T, theoretical TON (max) = 100), which veryfies catalytic activity but does not rival enzymatic reactions.12,24,30–33 However, considering the so far reported low catalytic activities of peptide- and protein-templated amidations,14–17 this represents a major advancement. Notably, observed rate accelerations are mainly caused by increasing the local concentration of reactants, rather than direct activation of functional groups, which distinguishes this system from most enzymatic reactions. Obtained activities are in line with early examples of nucleic-acid templated reactions which were later improved by adjusting reactant affinities and functional group alignments. The catalytic system introduced herein can serve as model system to investigate parameters crucial for protein-catalysed amidation, and it further expands the toolbox for selective protein modifications.
T. N. G. thanks the Fonds der Chemischen Industrie and the German Research Foundation (DFG, Emmy Noether program GR3592/2-1) for financial support. This work was supported by AstraZeneca, Bayer Crop Science, Bayer HealthCare, Boehringer Ingelheim, Merck KGaA, and the Max Planck Society. We thank Gernot Hahne for assistance with plasmid cloning.
Conflicts of interest
There are no conflicts to declare.References
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- B. List, Chem. Rev., 2007, 107, 5413–5415 CrossRef CAS.
- E. Roduner, Chem. Soc. Rev., 2014, 43, 8226–8239 RSC.
- D. Yi, T. Bayer, C. P. S. Badenhorst, S. Wu, M. Doerr, M. Höhne and U. T. Bornscheuer, Chem. Soc. Rev., 2021, 50, 8003–8049 RSC.
- R. Sterner and F. X. Schmid, Science, 2004, 304, 1916–1917 CrossRef CAS PubMed.
- P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem., 2018, 2, 409–421 CrossRef.
- A. Broom, R. V. Rakotoharisoa, M. C. Thompson, N. Zarifi, E. Nguyen, N. Mukhametzhanov, L. Liu, J. S. Fraser and R. A. Chica, Nat. Commun., 2020, 11, 4808 CrossRef CAS PubMed.
- M. Keller, E. Blöchl, G. Wächtershäuser and K. O. Stetter, Nature, 1994, 368, 836–838 CrossRef CAS PubMed.
- M. Frenkel-Pinter, M. Samanta, G. Ashkenasy and L. J. Leman, Chem. Rev., 2020, 120, 4707–4765 CrossRef CAS PubMed.
- T. Bose, G. Fridkin, C. Davidovich, M. Krupkin, N. Dinger, A. H. Falkovich, Y. Peleg, I. Agmon, A. Bashan and A. Yonath, Nucleic Acids Res., 2022, 50, 1815–1828 CrossRef CAS PubMed.
- A. Goswami and S. G. van Lanen, Mol. BioSyst., 2015, 11, 338–353 RSC.
- M. Schmidt, A. Toplak, P. J. L. M. Quaedflieg and T. Nuijens, Curr. Opin. Chem. Biol., 2017, 38, 1–7 CrossRef CAS PubMed.
- M. R. Petchey and G. Grogan, Adv. Synth. Catal., 2019, 361, 3895–3914 CrossRef CAS.
- D. H. Lee, J. R. Granja, J. A. Martinez, K. Severin and M. R. Ghadiri, Nature, 1996, 382, 525–528 CrossRef CAS PubMed.
- K. Severin, D. H. Lee, A. J. Kennan and M. Reza Ghadiri, Nature, 1997, 389, 706–709 CrossRef CAS PubMed.
- D. H. Lee, K. Severin, Y. Yokobayashi and M. R. Ghadiri, Nature, 1997, 390, 591–594 CrossRef CAS PubMed.
- A. Osuna Gálvez and J. W. Bode, J. Am. Chem. Soc., 2019, 141, 8721–8726 CrossRef PubMed.
- N. Brauckhoff, G. Hahne, J. T. H. Yeh and T. N. Grossmann, Angew. Chem., Int. Ed., 2014, 53, 4337–4340 CrossRef CAS PubMed.
- N. Vo and R. H. Goodman, J. Biol. Chem., 2001, 276, 13505–13508 CrossRef CAS PubMed.
- S. Brüschweiler, R. Konrat and M. Tollinger, ACS Chem. Biol., 2013, 8, 1600–1610 CrossRef PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- G. S. Beligere and P. E. Dawson, J. Am. Chem. Soc., 1999, 121, 6332–6333 CrossRef CAS.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed.
- T. N. Grossmann and O. Seitz, J. Am. Chem. Soc., 2006, 128, 15596–15597 CrossRef CAS PubMed.
- A. Erben, T. N. Grossmann and O. Seitz, Angew. Chem., Int. Ed., 2011, 50, 2828–2832 CrossRef CAS PubMed.
- W. C. Chan and P. D. White, Fmoc solid phase peptide synthesis: a practical approach, Oxford University Press, New York, 2000 Search PubMed.
- J. M. Goldberg, S. Batjargal, B. S. Chen and E. J. Petersson, J. Am. Chem. Soc., 2013, 135, 18651–18658 CrossRef CAS PubMed.
- M. Boudart, Chem. Rev., 1995, 95, 661–666 CrossRef CAS.
- D. S. Waugh, Protein Expression Purif., 2011, 80, 283–293 CrossRef CAS PubMed.
- D. Sievers and G. von Kiedrowski, Nature, 1994, 369, 221–224 CrossRef CAS PubMed.
- S. Sando, H. Abe and E. T. Kool, J. Am. Chem. Soc., 2004, 126, 1081–1087 CrossRef CAS PubMed.
- Z. L. Pianowski and N. Winssinger, Chem. Commun., 2007, 3820–3822 RSC.
- T. N. Grossmann, A. Strohbach and O. Seitz, ChemBioChem, 2008, 9, 2185–2192 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc00514c |
| This journal is © The Royal Society of Chemistry 2023 |