
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFast and slow walking driven by chemical fuel†
Vishnu Verman
Rajasekaran
 ,
Emad
Elramadi
,
Isa
Valiyev
,
Prodip
Howlader
and
Michael
Schmittel
*
,
Emad
Elramadi
,
Isa
Valiyev
,
Prodip
Howlader
and
Michael
Schmittel
*
Center of Micro and Nanochemistry and (Bio)Technology, Organische Chemie I, School of Science and Technology, University of Siegen, Adolf-Reichwein-Str. 2, D-57068, Germany. E-mail: schmittel@chemie.uni-siegen.de; Tel: +49(0) 2717404356
First published on 14th March 2023
Abstract
We demonstrate the fast forward and slow backward motion of a biped on a tetrahedral track using chemical fuel, cooperative binding and kinetic selectivity. Walking of the biped is based on its dibenzyl amine feet that bind to zinc porphyrin units and, upon protonation, to dibenzo 24-crown-8 sites affording pseudorotaxane linkages.
The myosin, dynein and kinesin family of motor proteins perform sophisticated, repetitive transport tasks essential to cellular functioning.1 Importantly, these intracellular transport tasks are performed out-of-equilibrium2 by the consumption of ATP3 as fuel. These complex, biological processes have instigated the design of both DNA-based4 as well as synthetic/semi-synthetic small-molecule walkers,5–8 often driven by chemical, light or biological inputs.
Molecular walkers have also been applied as transducers for catalytic reactions9 or cargo transport.4,10 However, most synthetic walkers rely on either strong covalent or kinetically inert coordination bonds, which firmly hold the biped on track. The desired sequential multi-step walking is then realized via chemical/light/semi-biological stimulation.11–13 In contrast, the concept of a multi-component walker held on track by dynamic interactions remains a largely unexplored area,14,15 and even more, if the system is chemically fueled.
In this report, we demonstrate how a multi-component assembly walks across a deck by exploiting the reversible switching between the NHamine → zinc-porphyrin16 (= NHamine → ZnPor) and dibenzylammonium pseudorotaxane binding17 motifs. The process may be started with acid and reversed with base, or in a pulsed forward–backward motion using a fuel acid,18 for instance 319 (Fig. 1). Upon protonation of [1•2] the dibenzylamine sites are protonated and insert as secondary ammonium units into the dibenzo-24-crown-8 binding sites of deck 1 affording two [2]pseudorotaxane linkages in [1•2(H+)2]. With base the walking is reversed.
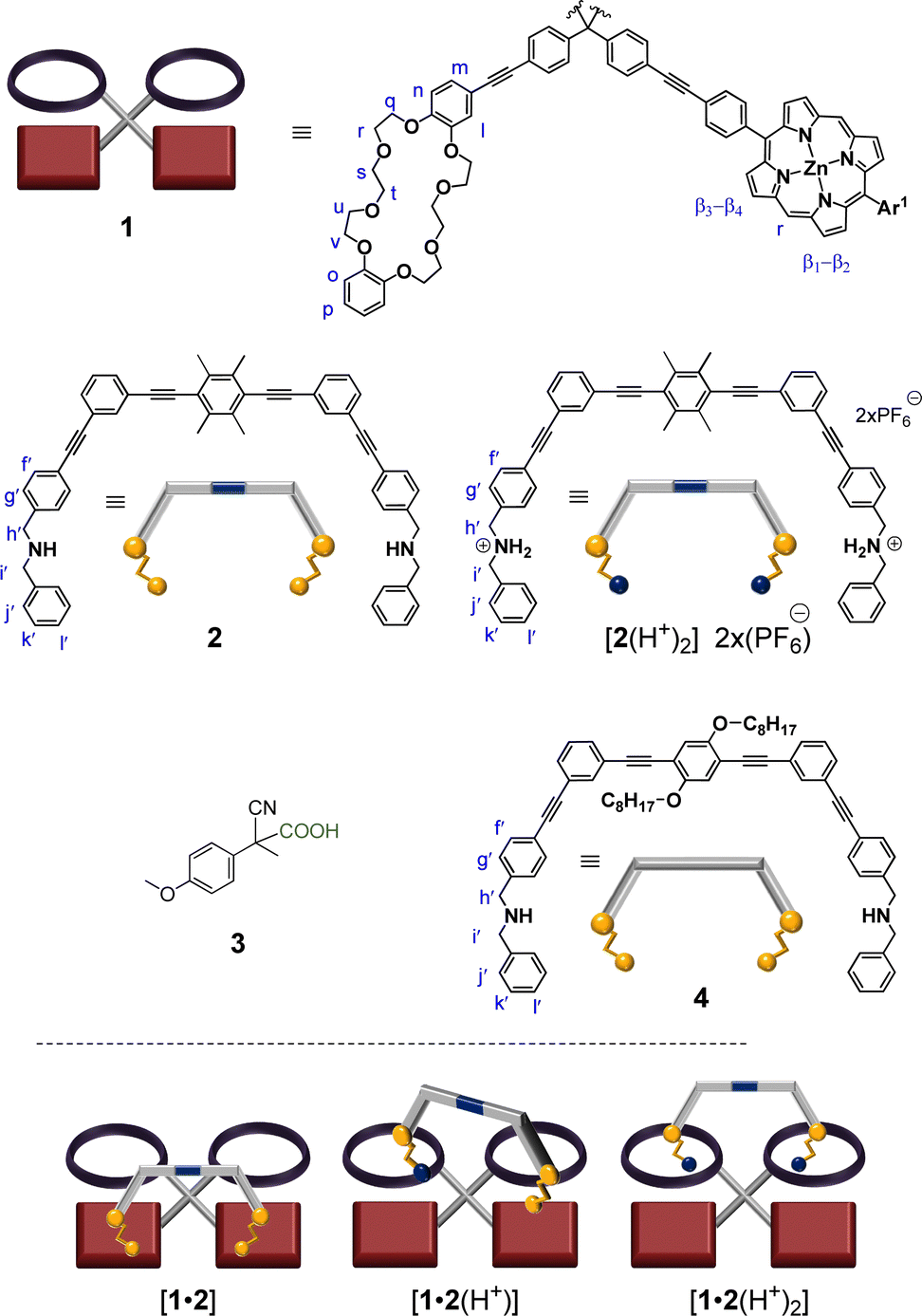 | ||
| Fig. 1 Ligands used in this study. (The cartoon version of the tetrahedral deck 1 is a 2D-projection). | ||
The design strategy for the walker-on-deck was based on recent reports from our group, where we demonstrated that NPy → ZnPor and pseudorotaxane linkages are orthogonal interactions.20 The synthesis and characterization of deck 1 and other ligands used in this study (Fig. 1) are described in detail in the ESI.† Upon mixing deck 1 and biped 2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the assembly [1•2] was quantitatively afforded. The walker-on-deck was fully characterized by 1H, 1H-1H COSY and elemental analysis (ESI,† Fig. S21, S22 and S29). Further, the 1H DOSY of [1•2] yielded a single, sharp trace with a D = 4.40 × 10−10 m2 s−1 confirming the quantitative complexation between 1 and 2 (ESI,† Fig. S29). Foremost, [1•2] was identified by the changes in the 1H-NMR signals of protons j′-H and g′-H of biped 2 as walker and those of proton r-H of deck 1 (Fig. 2c–e). The stark changes of the j′-H and g′-H signals were attributed to interaction with the porphyrin's shielding ring current upon axial NHamine → ZnPor coordination, which consequently shifted the r-H proton signals of 1 upfield.
:1), the assembly [1•2] was quantitatively afforded. The walker-on-deck was fully characterized by 1H, 1H-1H COSY and elemental analysis (ESI,† Fig. S21, S22 and S29). Further, the 1H DOSY of [1•2] yielded a single, sharp trace with a D = 4.40 × 10−10 m2 s−1 confirming the quantitative complexation between 1 and 2 (ESI,† Fig. S29). Foremost, [1•2] was identified by the changes in the 1H-NMR signals of protons j′-H and g′-H of biped 2 as walker and those of proton r-H of deck 1 (Fig. 2c–e). The stark changes of the j′-H and g′-H signals were attributed to interaction with the porphyrin's shielding ring current upon axial NHamine → ZnPor coordination, which consequently shifted the r-H proton signals of 1 upfield.
 | ||
| Fig. 2 Partial comparison of 1H-NMR (500 MHz, CD2Cl2, 298 K) of (a) free biped [2(H+)2](PF6−)2, (b) [2]pseudorotaxane [1•2(H+)2](PF6−)2, (c) free deck 1, (d) [1•2], and (e) free biped 2. | ||
Similarly, 1 and 2 were mixed with TFA in a 1:1:2 ratio to assemble [1•2(H+)2]. The double protonation of 2, however, in parallel removed Zn2+ (ca. 50%) from the ZnPor sites of deck 1. Instead, we achieved [2]pseudorotaxane formation with HPF6 as acid by first generating [2(H+)2](PF6−)2, which in a second step was combined with deck 1 (Fig. 2a and b).
Complex [1•2(H+)2](PF6−)2 was fully characterized by 1H, 1H-1H COSY NMR and elemental analysis. Additionally, the 1H DOSY of [1•2(H+)2](PF6−)2 yielded a single, sharp trace with D = 3.30 × 10−10 m2 s−1 confirming quantitative complexation between 1 and [2(H+)2](PF6−)2 (ESI,† Fig. S28). Most diagnostic were the changes in the 1H-NMR signature of proton signals o-H, p-H of 1 and h′-H, i′-H adjacent to the ammonium units of [1•2(H+)2]. The proton signals of o-H & p-H split into a complex multiplet due to the de-symmetrization induced by threading. The proton signals h′-H & i′-H of [1•2(H+)2] (4.27–4.23 ppm in [2(H+)2](PF6−)2) shifted downfield to 4.74 ppm (broad) in [1•2(H+)2] after [2]pseudorotaxane formation (Fig. 2a and b).
Before studying the dissipative system, we tested the stepwise deprotonation-driven walking using DBU as base (Scheme 1). Pseudorotaxane [1•2(H+)2](PF6−)2 was titrated with DBU (increments of 0.1 equiv.) using the proton r-H signal as an indicator (Fig. 3). Upon addition of DBU, the proton r-H signal split into three sets: two sets from [1•2(H+)] with two distinct ZnPor units (unbound: 10.40 ppm; bound: 10.36 ppm) and one set from [1•2(H+)2] with two degenerate ZnPor sites (10.41 ppm). From 0.1–2.0 equiv. of DBU, the integration of the r-H signal at 10.36 ppm (w.r.t. to an internal standard) furnished the amount of [1•2(H+)] in the mixture of [1•2(H+)2], [1•2(H+)], and [1•2] as shown in Fig. 3a. Further, a plot of the percentage of [1•2(H+)] vs. the equiv. of added DBU revealed some interesting findings: First, there is a steady growth (from 0 to 86%) of [1•2(H+)] during the addition of 0.1–1.2 equiv. of DBU followed by its decay to form [1•2]. Secondly, the plot clearly shows that at least 86% (peak of the inverted parabolic-fit) of the back walking of [1•2(H+)2] → [1•2] occurs stepwise by mono-deprotonation. After adding 1.0 equiv. of DBU (ESI,† Fig. S31), the mixture is composed of [1•2(H+)] (80%) and [1•2(H+)2] (20%). After 1.4–2.0 equiv. of DBU had been added, the diagnostic j′, g′-H proton signals of [1•2] around 6.7 ppm appeared, adding further support to the stepwise deprotonation pathway.
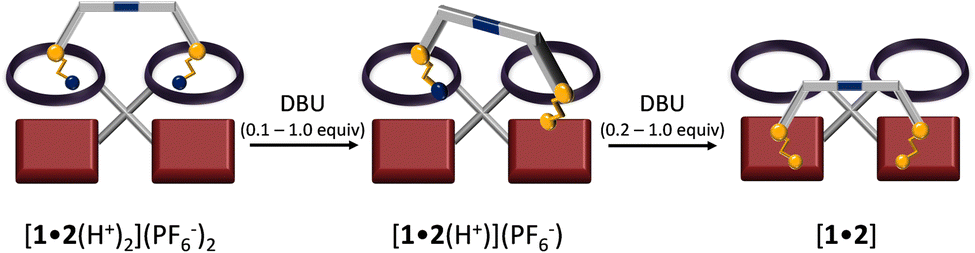 | ||
| Scheme 1 Stepwise walking of biped 2 on deck 1 by step-by-step deprotonation of the bis-pseudorotaxane [1•2(H+)2](PF6−)2 by titration with DBU. The intermediately formed [1•2(H+)](PF6−) then undergoes gradual deprotonation to the di-NHAmine → ZnPor bound complex [1•2]. | ||
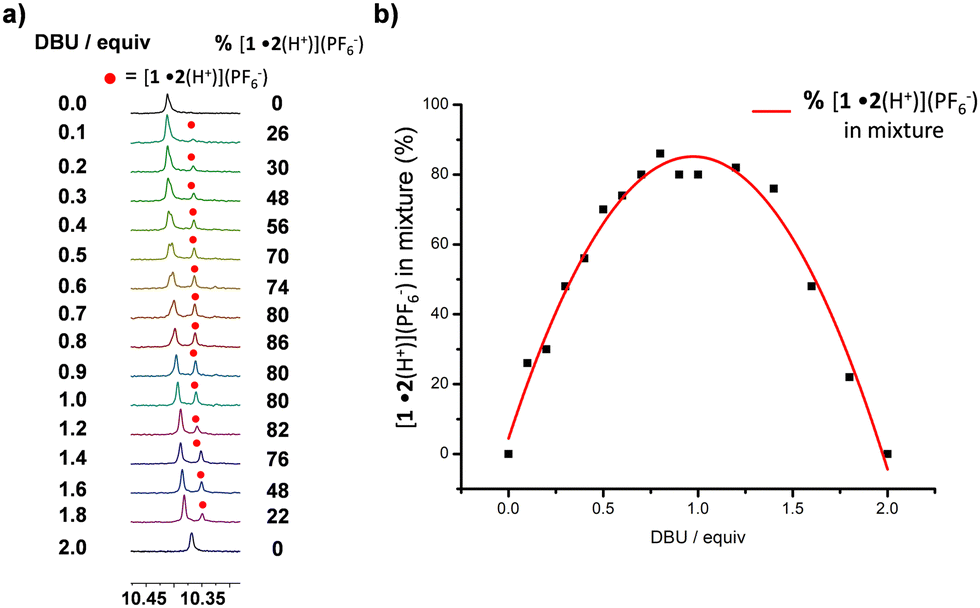 | ||
| Fig. 3 (a) Partial 1H-NMR stack showing the changes in the proton r-H signal upon titration of [1•2(H+)2](PF6−)2 with DBU. (b) Percentage of [1•2(H+)](PF6−) in the mixture against equiv. of DBU added w.r.t to the initial assembly [1•2(H+)2](PF6−)2. | ||
 | ||
| Scheme 2 Chemically fueled switching between [1•4] and the bis-[2]pseudorotaxane [1•4(H+)2] using the fuel acid 3. | ||
One would expect the monoprotonated [1•2(H+)] to undergo sliding of the amine foot between both zinc porphyrin sites and/or the ammonium foot between the two degenerate dibenzo24-crown-8 units. Surprisingly, a 1H-1H ROESY-NMR using the o,p-H and r-H as diagnostic signals revealed exchange peaks neither between the two crown ethers nor between both zinc porphyrin units (ESI,† Figure S27). Accordingly, the amine foot did not exchange between the two zinc porphyrins and by extension there was no chemical exchange between the two distinct r-H signals in [1•2(H+)]. Thus, the biped takes a steady walking motion from [1•2(H+)2] through [1•2(H+)] to [1•2].
With these findings at hand, we decided to study the operation of dissipative walking using fuel 3. Unexpectedly, ligand 2 was not suitable for this purpose as [2(H+)2] proved to be insoluble. To remedy this problem, ligand 4 was designed as an analog of 2 with bis-octoxy chains for higher solubility. Consequently, now ligands 1, 4 were mixed to quantitatively furnish [1•4] which was fully characterized by 1H, 1H-1H COSY, 1H DOSY (D = 3.50 × 10−10 m2 s−1) and elemental analysis. (ESI,† Fig. S25, S26 and S30) Subsequently, 2.5 equiv. of fuel acid 3 was added for conversion of [1•4] to [1•4(H+)2] (Scheme 2). 1H-NMR indicated the disappearance of the drastically upfield shifted broad signals of protons j′-H, g′-H in [1•4] along with a shift of the r-H proton from 10.37 to 10.39 ppm as in the free deck 1 (Fig. 4). Proton signals j′,g′-H shifted from 6.65 and 6.58 ppm in [1•4] to 7.42 and 7.28 ppm in [1•4(H+)2], respectively. Moreover, proton signals o,p-H of deck 1 showed an intricate splitting into two sets similar to those in [1•2(H+)2], which supported formation of bis-[2]pseudorotaxane [1•4(H+)2].
 | ||
| Fig. 4 Comparison of partial 1H-NMR spectra as a function of time (500 MHz, CD2Cl2, 298 K) during the first fueled motion cycle. | ||
The stepwise walking from [1•4(H+)2] back to [1•4] via step-by-step deprotonation was also monitored by 1H-NMR. The di-threaded bis-[2]pseudorotaxane [1•4(H+)2] prevailed in the mixture from t = 8 to 80 min as evidenced by the single r-H signal at 10.39 ppm along with the split o,p-H signals (Fig. 4: Phase 1). From t = 80 to 125 min we clearly see the emergence of two distinct sets of r-H signals (10.39 and 10.38 ppm) also accompanied by shifts of the β2, β3-H signals in the range of 9.4–9.6 ppm (Phase 2 in Fig. 4). The r-H signals at 10.39 and 10.38 ppm correspond to the di-threaded [1•4(H+)2] and mono-threaded [1•4(H+)], respectively. At time t = 115–130 min, the full conversion of [1•4(H+)2] to the monoprotonated [1•4(H+)] was finished, as indicated by nearly complete disappearance of [1•4(H+)2] (10.39 ppm) and simultaneous increase in the signal corresponding to [1•4(H+)] (10.38 ppm). Alike, the β-protons of [1•4(H+)2] disappeared while the signals from [1•4(H+)] simultaneously increased showing the same stepwise walking protocol.
From t = 130 to 230 min, we then observe the complete conversion of the mixture into [1•4] (Phase 3 in Fig. 4) due to the second deprotonation. The gradual emergence of the h′, i′-H signals (2.55–2.65 ppm) (Fig. S33, ESI†) and g′, j′-H signals (6.64–6.71 ppm) (Phase 3 in Fig. 4) are attributed to the reformation of [1•4]. Although, fuel 3 has been utilized to drive several dissipative networks21–23 and a standalone molecular rotor,24 this report represents the first use of 3 to drive a walker-on-deck. The reproducibility of the walking process was tested twice by running two consecutive fueled walking cycles with minimal disruption by the waste products from the chemical fuel consumption apart from a slightly longer 2nd cycle operation time (362 min vs. 230 min) (Fig. 5a and b). From the data, we recognize that walking from [1•4] to [1•4(H+)2] is fast, while the return via the intermediate [1•4(H+)] is slow, as if a brake pad25 would apply. For functional utility in the future this opens the opportunity to take up a load in the long-lived [1•4(H+)2] and carry it to [1•4].
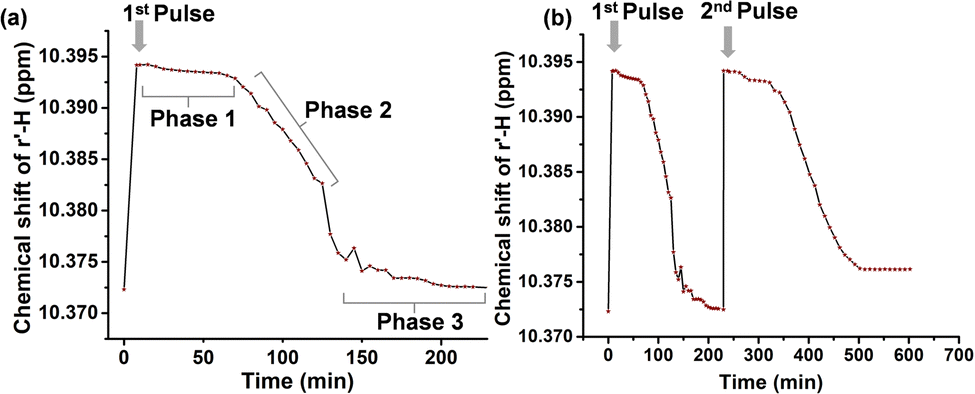 | ||
| Fig. 5 (a) Plot of chemical shift of r-H of 1versus time during the first cycle from [1•4] to [1•4(H+)2] and back. The three phases in the transition (Phases 1–3) are populated by different proportions of the assemblies. (b) Plot of chemical shift of proton r-H of 1versus time over two fueled cycles monitored by 1H-NMR. The biped 4 was twice driven from [1•4] to [1•4(H+)2] and automatically back to [1•4] with minimal degradation in function. | ||
In summary, we have demonstrated unprecedented reversible and stepwise walking of a small biped on a tetrahedral track using a fuel acid. Further, the three-step walking motion is based on a novel, pulsed switching protocol between orthogonally self-sorted NHAmine → ZnPor and ammonium ⊂ crown-ether pseudorotaxane interactions. The combination of multiple self-sorting26,27 and fueling emphasizes the potential of leveraging dissipative processes28–30 to achieve sophisticated functions in multi-component, dynamic supramolecular assemblies.
We are indebted to the University of Siegen and the Deutsche Forschungsgemeinschaft (Schm 647/22-1) for continued support.
Conflicts of interest
There are no conflicts to declare.References
- (a) R. D. Vale and R. A. Milligan, Science, 2000, 288, 88 CrossRef CAS; (b) R. D. Vale, Cell, 2003, 112, 467–480 CrossRef CAS; (c) M. Schliwa and G. Woehlke, Nature, 2003, 422, 759–765 CrossRef CAS.
- S. De and R. Klajn, Adv. Mater., 2018, 30, e1706750 CrossRef.
- M. A. Cardona, R. Chen, S. Maiti, I. Fortunati, C. Ferrante, L. Gabrielli, K. Das and L. J. Prins, Chem. Commun., 2020, 56, 13979–13982 RSC.
- J. S. Shin and N. A. Pierce, J. Am. Chem. Soc., 2004, 126, 10834–10835 CrossRef CAS.
- M. von Delius and D. A. Leigh, Chem. Soc. Rev., 2011, 40, 3656–3676 RSC.
- D. H. Qu and H. Tian, Chem. Sci., 2013, 4, 3031–3035 RSC.
- C. J. Martin, A. T. L. Lee, R. W. Adams and D. A. Leigh, J. Am. Chem. Soc., 2017, 139, 11998–112002 CrossRef CAS PubMed.
- Z. Wang, R. Hou and I. Y. Loh, Nanoscale, 2019, 11, 9240–9263 RSC.
- N. Mittal, M. S. Özer and M. Schmittel, Inorg. Chem., 2018, 57, 3579–3586 CrossRef CAS PubMed.
- L. Zheng, H. Zhao, Y. Han, H. Qian, L. Vukovic, J. Mecinović, P. Král and W. T. S. Huck, Nat. Chem., 2019, 11, 359–366 CrossRef CAS PubMed.
- M. J. Barrell, A. G. Campaña, M. von Delius, E. M. Geertsema and D. A. Leigh, Angew. Chem., Int. Ed., 2011, 50, 285–290 CrossRef CAS PubMed.
- J. E. Beves, V. Blanco, B. A. Blight, R. Carrillo, D. M. D’Souza, D. Howgego, D. A. Leigh, A. M. Z. Slawin and M. D. Symes, J. Am. Chem. Soc., 2014, 136, 2094–2100 CrossRef CAS PubMed.
- C. J. Martin, A. T. L. Lee, R. W. Adams and D. A. Leigh, J. Am. Chem. Soc., 2017, 139, 11998–112002 CrossRef CAS PubMed.
- A. G. Campaña, A. Carlone, K. Chen, D. T. F. Dryden, D. A. Leigh, U. Lewandowska and K. M. Mullen, Angew. Chem., Int. Ed., 2012, 51, 5480–5483 CrossRef PubMed.
- P. Kovaříček and J. M. Lehn, Chem. – Eur. J., 2015, 21, 9380–9384 CrossRef.
- L. Favereau, A. Cnossen, J. B. Kelber, J. Q. Gong, R. M. Oetterli, J. Cremers, L. M. Herz and H. L. Anderson, J. Am. Chem. Soc., 2015, 137, 14256–14259 CrossRef CAS.
- K. Nowosinski, L. K. S. von Krbek, N. L. Traulsen and C. A. Schalley, Org. Lett., 2015, 17, 5076–5079 CrossRef CAS PubMed.
- E. Olivieri and A. Quintard, ACS Org. Inorg. Au, 2023, 3, 4–12 CrossRef CAS.
- (a) C. Biagini and S. Di Stefano, Angew. Chem., Int. Ed., 2020, 59, 8344–8354 CrossRef CAS; (b) J. A. Berrocal, C. Biagini, L. Mandolini and S. Di Stefano, Angew. Chem., Int. Ed., 2016, 55, 6997–7001 CrossRef CAS; (c) C. Biagini, S. Albano, R. Caruso, L. Mandolini, J. A. Berrocal and S. Di Stefano, Chem. Sci., 2018, 9, 181–188 RSC.
- A. Ghosh, I. Paul and M. Schmittel, J. Am. Chem. Soc., 2021, 143, 5319–5323 CrossRef CAS.
- A. Ghosh, I. Paul and M. Schmittel, J. Am. Chem. Soc., 2019, 141, 18954–18957 CrossRef CAS.
- D. Mondal, A. Ghosh, I. Paul and M. Schmittel, Org. Lett., 2022, 24, 69–73 CrossRef CAS.
- I. Valiyev, A. Ghosh, I. Paul and M. Schmittel, Chem. Commun., 2022, 58, 1728–1731 RSC.
- A. Goswami, S. Saha, E. Elramadi, A. Ghosh and M. Schmittel, J. Am. Chem. Soc., 2021, 143, 14926–14935 CrossRef CAS PubMed.
- E. Elramadi, A. Ghosh, I. Valiyev, P. K. Biswas, T. Paululat and M. Schmittel, Chem. Commun., 2022, 58, 8073–8076 RSC.
- Z. He, W. Jiang and C. A. Schalley, Chem. Soc. Rev., 2015, 44, 779–789 RSC.
- S. De, S. Pramanik and M. Schmittel, Angew. Chem., Int. Ed., 2014, 53, 14255–14259 CrossRef CAS PubMed.
- (a) K. Das, L. Gabrielli and L. J. Prins, Chemically Fueled Self-Assembly in Biology and Chemistry, Angew. Chem., Int. Ed., 2021, 60, 20120–20143 CrossRef CAS PubMed; (b) Q. Wang, Z. Qi, M. Chen and D. Qu, Aggregate, 2021, 2, e110 CAS.
- V. W. Liyana Gunawardana, T. J. Finnegan, C. E. Ward, C. E. Moore and J. D. Badjić, Angew. Chem., Int. Ed., 2022, 61, e202207418 CrossRef CAS PubMed.
- D. Del Giudice, M. Valentini, G. Melchiorre, E. Spatola and S. Di Stefano, Chem. – Eur. J., 2022, 28, e202200685 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: synthesis, spectroscopic characterization, mechanistic and fuel studies. See DOI: https://doi.org/10.1039/d3cc00357d |
| This journal is © The Royal Society of Chemistry 2023 |