
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface modification of gold by carbazole dendrimers for improved carbon dioxide electroreduction†
Sota
Yoshida
a,
Masaki
Sampei
a,
Naoto
Todoroki
 *ab,
Eri
Hisamura
c,
Kohei
Nakao
c,
Ken
Albrecht
bc and
Toshimasa
Wadayama
a
*ab,
Eri
Hisamura
c,
Kohei
Nakao
c,
Ken
Albrecht
bc and
Toshimasa
Wadayama
a
aGraduate School of Environmental Studies, Tohoku University, 6-2-2 Aramakiaza-Aoba Aoba-ku, Sendai 980-8579, Japan. E-mail: naoto.todoroki.b1@tohoku.ac.jp
bJapan Science and Technology Agency, PRESTO, Kawaguchi, Saitama 332-0012, Japan
cInstitute for Materials Chemistry and Engineering, Kyushu University, 6-1 Kasuga-koen, Kasuga-shi, Fukuoka 816-8580, Japan
First published on 24th February 2023
Abstract
Four types of carbazole dendrimers were applied as modification molecules of Au surfaces to improve carbon dioxide electroreduction. The reduction properties depended on the molecular structures: the highest activity and selectivity to CO was achieved by 9-phenylcarbazole, probably caused by the charge transfer from the molecule to Au.
Practical carbon dioxide conversion processes are being considered to generate valuable chemicals for achieving carbon neutrality.1 Electrochemical CO2 reduction is crucial as one of the conversion processes of CO2 to carbon monoxide (CO) and various chemicals such as hydrocarbons, alcohols, and formic acid, at ambient temperature and pressure. Therefore, practical electrolysis systems and the required materials have been intensively investigated.2–7 Previous research has shown that Au, Ag, and Zn electrodes selectively convert CO2 to CO.8 In particular, Au showed relatively high efficiency for CO2 reduction compared to that for hydrogen evolution, which is a competitive reaction of CO2 reduction in aqueous solutions.8–10 However, the activity and selectivity of the Au electrode for converting CO2 to CO remain insufficient for widespread application, and further research on CO2 electrolysis systems is necessary to attain industrialisation.
We should notice that Au electrodes modified with various organic molecules enhance the reduction activity and selectivity to CO.11–19 For example, Zhao et al. used various amines to modify the surfaces of Au nanoparticles and found that oleylamine effectively improved the selectivity to CO.11 Lee et al. investigated surface-modified Au nanoparticles experimentally and theoretically using density functional theory calculations, where polymeric binders such as Nafion, polyvinyl alcohols, and polytetrafluoroethylene (PTFE) were used as modifying molecules. They showed that PTFE improved the selectivity to CO and the enhanced selectivity was originated from promoting CO2 adsorption on the surface and suppressing the competing hydrogen evolution reaction (HER).20 These reports clearly indicate that surface-modifying molecules affect the adsorption of CO2 and H2O on Au electrode surfaces and determine the efficiency of the electrochemical CO2 reduction reaction (CO2RR).
Dendrimers are dendritic polymers with precisely controlled structures that have potential applications as electronic and catalytic materials.21–23 Carbazole dendrimers (CDs), in particular, have unique photo- and electrochemical properties, and therefore are applicable as hole transport and luminescent materials.18,24 The molecular backbones, functional groups, and molecular weights of CDs can be easily controlled as a function of the generation (the number of repeated branching cycles). Furthermore, the chemical and physical properties of CDs can be tuned by the terminal and core functional groups. Due to molecular designability and resulting unique properties, CDs are also applicable for fundamental study to understand the relationship between the surface modifications of electrode surfaces.
In this study, four types of CDs were used to modify Au electrode surface for CO2 electrochemical reduction. First, we showed that the conversion of CO2 to CO using an Au disk electrode in an H-type electrochemical cell was enhanced most by the 9-phenylcarbazole (G1Ph) modification. Then, the enhanced electrochemical CO2 reduction was also confirmed at high current density electrolysis using a gas diffusion electrode (GDE) cell designed for use in a practical CO2 electrolyser.
A CD-modified polycrystalline Au disk (ϕ = 5 mm) was used as the working electrode for CO2 electrochemical reduction experiments in a CO2-saturated 0.1 M KHCO3 solution using an H-type electrochemical cell. The four CDs investigated are depicted in Fig. 1. The CDs with phenyl (Ph) or NH groups are described as GxPh (x = generation number of 1 and 3) and GxCz, respectively. The surface modification by the CDs was conducted by drop-casting toluene solutions containing 0.1 mM CDs onto the Au disk electrode, followed by drying in air.
 | ||
| Fig. 1 Molecular structures of the carbazole dendrimers used for surface modification of the Au electrode. | ||
Fig. 2 shows the CO2 reduction properties of the CD-modified and non-modified Au electrodes. In this condition, CO was the only product of the CO2RR, irrespective of the kinds of CDs, and thus faradaic efficiencies and partial current densities for the generated CO are estimated (described in detail in supplementary information; ESI†). As shown in Fig. 2(a), the G1Ph-modified electrode showed enhanced faradaic efficiencies (FECO) relative to the non-modified electrode at −0.4, −0.5, and −0.6 V vs. reversible hydrogen electrode (RHE), particularly ca. 10% higher FECO at −0.4 V. In contrast, the G1Cz-, G3Cz-, and G3Ph-modified electrodes exhibited enhanced FECO at −0.4 V, while the efficiencies at −0.5 V and −0.6 V were almost identical to or lower than that of non-modified Au.
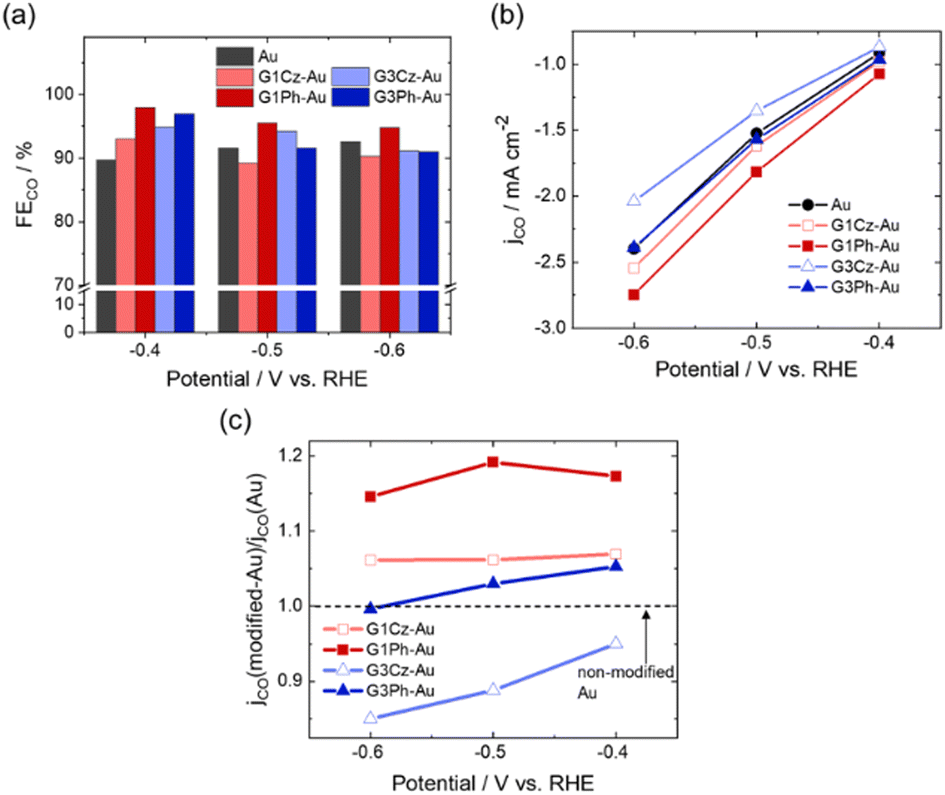 | ||
| Fig. 2 (a) Faradaic efficiency and (b) partial current density for CO of surface-modified and non-modified Au electrodes evaluated in a CO2-saturated 0.1 M KHCO3 solution. (c) Normalized CO partial current density of G1Ph, G3Ph- and G1Cz, G3Cz-modified Au. | ||
Fig. 2(b) and (c) show the partial current densities for CO (jCO) of the CD-modified electrode surfaces, where the current densities are normalised to that of the non-modified Au electrode at each applied potential. As shown in (b), G1Ph- and G1Cz-modified Au electrodes exhibited enhanced jCO, while the current densities of the G3Cz- and G3Ph-modified electrodes were comparable to and less than that of the non-modified electrode, respectively. These results suggest that CDs of smaller size (G1Ph and G1Cz) were favourable surface-modifying molecules. The maximum enhancement factor of jCO was ca. 1.2 for G1Ph at −0.5 V, in comparison to non-modified Au (c). The results clearly indicate that introducing a phenyl group to the first generation carbazole (G1Ph) was the most effective surface modification at the applied potentials tested (−0.4 to −0.6 V).
Pb-underpotential deposition (UPD) was conducted for the CD-modified Au electrodes to estimate the active surface areas for the CO2RR. Fig. 3(a) shows cyclic voltammograms recorded in an Ar-purged 1 mM Pb(OAc)2 + 0.1 M NaOH solution for the most effective G1Ph-modified Au, less-effective G3Ph-modified, and non-modified Au electrodes. Because Pb-UPD potential is sensitive to the surface crystal facets of Au electrodes,25 active surface areas of CD-modified Au electrode surfaces can be estimated for each crystal face of the modified Au electrodes. The potential region below 0.3 V vs. RHE (green) corresponds to the overpotential deposition (OPD) of Pb. The peaks located at the approximately 0.34 V (blue) and 0.51 V (red) regions in an anodic sweep can be ascribed to the stripping of Pb from the (111) and (110) facets, respectively, of the non-modified Au (black).25 According to a previous report,25 the Pb-stripping peak for the Au(100) face appears at ca. 0.4 V. In contrast, the corresponding peak is absent in Fig. 3(a), suggesting that the Au polycrystalline electrode used in this study was mainly composed of the (111) and (110) facets.
 | ||
| Fig. 3 (a) Cyclic voltammograms for Pb underpotential deposition collected in an Ar-purged 1 mM Pb(OAc)2 + 0.1 M NaOH solution. (b) Pb-stripping charges estimated from (a). XP spectra of N 1s (c) and Au 4f (d) orbitals for G1Ph-modified and non-modified Au. (e) Linear-sweep voltammograms for the HER recorded in Ar-purged 0.1 M PBS electrolyte with a pH of 6.8. | ||
The Pb-striping peaks for G1Ph- (red) and G3Ph-modified (blue) Au electrodes are located at similar potential regions for the non-modified one (black), suggesting that the (110) and (111) facets of the Au electrode surfaces are even active for CO2 reduction sites under the presence of modifying molecules, though the estimated average Pb-stripping charges slightly decreased, particularly for the (110) facet, even by the effective G1Ph-surface-modification (Fig. 3(b)).
Furthermore, the Pb-stripping peaks shifted to approximately 20 mV higher potential for G1Ph-Au compared to the non-modified electrode. The positive potential shift suggests that G1Ph modification changed the electronic properties of the Au surface. Indeed, a similar shift has been reported for porphyrin-modified Au nanoparticles.26 Modification with G3Ph caused a marked decrease in the corresponding charges, particularly in the (110) facet, suggesting that the lower efficiency might correspond to reaction-site blocking on the (110) facet.9 Therefore, the molecular-modification-dependent difference in Pb-stripping charge probably stems from differences in the molecular size of the CDs. The Pb-UPD experiment results suggest that G1Ph modification-enhanced CO2 electrochemical reduction property is originated from suitable surface electronic states on the modified Au electrode, since the active surface area was nearly unchanged by the surface modification. In contrast, the G3Ph-modification decreased the surface active area, probably as a consequence of the larger molecular sizes of the CDs.
The change in the surface electronic state by G1Ph modification was also investigated by X-ray photoelectron spectroscopy (XPS). Fig. 3(c) and (d) show the XP spectra of the N 1s and Au 4f orbitals of the G1Ph-modified and non-modified Au. In the N 1s band of G1Ph-Au, the peak at 400 eV can be attributed to the C–N bond in the five-membered ring of carbazole.27 Simultaneously, the Au 4f7/2 band of the G1Ph-Au shows a higher binding energy shift of ca. 0.2 eV, relative to that of non-modified Au. CDs have electron-donating properties,28 and thus G1Ph may donate electrons to the Au electrode surface.
To understand the influence of CD surface modification on the competing HER, linear-sweep voltammetry (LSV) was conducted for G1Ph-modified Au in an Ar-purged 0.1 M phosphate-buffered saline solution with pH of 6.8, i.e., water electrolysis at the same pH (Fig. 3(e)). The LSV curves of G1Ph- (red) and non-modified (black dashed) Au electrodes are similar to each other, suggesting that the G1Ph modification does not affect the HER on the Au electrode surfaces. Zhang et al. proposed that the elementary step of CO2 to a COOH* intermediate is the rate-limiting step for electrochemical CO2 reduction on Au surfaces.2 As shown previously for the Pb-UPD and XPS results (Fig. 3), the G1Ph-modification probably causes local electron enrichment at the Au electrode surface that can promote CO2 reduction by reducing the activation energy for the first reductive step of CO2 to COOH*.9,26
Finally, we confirmed the enhanced CO2 electrochemical reduction activity of the G1Ph-modified Au electrode at a high current density (∼100 mA cm−2). Fig. 4(a) presents a schematic of the flow-type GDE cell used in this study. The 100 nm-thick Au thin film deposited by magnetron sputtering (see ESI†) on a gas diffusion layer (Au-GDL) was used as a working electrode. Then, the Au-GDL was modified using a toluene solution of 0.1 mM G1Ph by the same procedure described for the Au disk electrode. Fig. 4(b) and (c) show the FECO and jCO of the G1Ph-(red) and non-modified (black) Au-GLD. As clearly shown, the estimated FECO and jCO increase ca. 10–20% by G1Ph modification, except for jCO at −0.5 V, indicating the feasibility of the G1Ph-modified Au electrode for practical CO2 electrochemical conversion to CO.
 | ||
| Fig. 4 (a) Schematic of the gas diffusion electrode type flow cell. (b) Faradaic efficiency and (c) partial current density for CO of G1Ph-modified and non-modified Au-GDL. | ||
In conclusion, we investigated the electrochemical CO2 reduction for CD-modified Au electrodes. The reduction property conducted by using Au disk electrodes and an H-type electrochemical cell depended on the backbone size (generation) and functional groups of the CDs. Among the CDs, G1Ph was most effective for CO2 conversion to CO. Based on the results of XPS, Pb-UPD, and HER experiments, the enhanced electrochemical CO2 reduction of the G1Ph-modified Au can be explained by a charge transfer from G1Ph to the Au surface. G1Ph-modification also enhanced high-current density electrolysis (∼100 mA cm−2) in a GDE electrochemical setup. These results show that the G1Ph surface modification of Au electrode surfaces can facilitate a practical electrochemical conversion of CO2 to CO.
This study was partly supported by JST, PRESTO Grand Number JPMJPR20T3 (N. T.) and JPMJPR18T2 (K. A.), Japan, and the Leading Initiative for Excellent Young Researchers from the Ministry of Education, Culture, Sport, Science & Technology of Japan (K. A.). The authors would also like to thank Dr. N. Akao and Dr. Y. Ohira for XPS measurements.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. Navarro-Jaén, M. Virginie, J. Bonin, M. Robert, R. Wojcieszak and A. Y. Khodakov, Nat. Rev. Chem., 2021, 5, 564–579 CrossRef.
- B. Zhang, Y. Jiang, M. Gao, T. Ma, W. Sun and H. Pan, Nano Energy, 2021, 80, 105504 CrossRef CAS.
- D. Higgins, C. Hahn, C. Xiang, T. F. Jaramillo and A. Z. Weber, ACS Energy Lett., 2019, 4, 317–324 CrossRef CAS.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- N. Todoroki, N. Yokota, S. Nakahata, H. Nakamura and T. Wadayama, Electrocatalysis, 2016, 7, 97–103 CrossRef CAS.
- K. Iwase, T. Kojima, N. Todoroki and I. Honma, Chem. Commun., 2022, 58, 4865–4868 RSC.
- I. E. L. Stephens, K. Chan, A. Bagger, S. W. Boettcher, J. Bonin, E. Boutin, A. K. Buckley, R. Buonsanti, E. R. Cave, X. Chang, S. W. Chee, A. H. M. da Silva, P. de Luna, O. Einsle, B. Endrődi, M. Escudero-Escribano, J. V. Ferreira de Araujo, M. C. Figueiredo, C. Hahn, K. U. Hansen, S. Haussener, S. Hunegnaw, Z. Huo, Y. J. Hwang, C. Janáky, B. S. Jayathilake, F. Jiao, Z. P. Jovanov, P. Karimi, M. T. M. Koper, K. P. Kuhl, W. H. Lee, Z. Liang, X. Liu, S. Ma, M. Ma, H.-S. Oh, M. Robert, B. R. Cuenya, J. Rossmeisl, C. Roy, M. P. Ryan, E. H. Sargent, P. Sebastián-Pascual, B. Seger, L. Steier, P. Strasser, A. S. Varela, R. E. Vos, X. Wang, B. Xu, H. Yadegari and Y. Zhou, JPhys Energy, 2022, 4, 042003 CrossRef.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- N. Todoroki, H. Tei, H. Tsurumaki, T. Miyakawa, T. Inoue and T. Wadayama, ACS Catal., 2019, 9, 1383–1388 CrossRef CAS.
- N. Todoroki, H. Tei, T. Miyakawa, H. Tsurumaki and T. Wadayama, ChemElectroChem, 2019, 6, 3101–3107 CrossRef CAS.
- Y. Zhao, C. Wang, Y. Liu, D. R. MacFarlane and G. G. Wallace, Adv. Energy Mater., 2018, 8, 1801400 CrossRef.
- J. Wang, J. Yu, M. Sun, L. Liao, Q. Zhang, L. Zhai, X. Zhou, L. Li, G. Wang, F. Meng, D. Shen, Z. Li, H. Bao, Y. Wang, J. Zhou, Y. Chen, W. Niu, B. Huang, L. Gu, C. S. Lee and Z. Fan, Small, 2022, 18, e2106766 CrossRef PubMed.
- Q. Lenne, Y. R. Leroux and C. Lagrost, ChemElectroChem, 2020, 7, 2345–2363 CrossRef CAS.
- J. Wang, F. Xu, Z. Y. Wang, S. Q. Zang and T. C. W. Mak, Angew. Chem., Int. Ed., 2022, 61, e202207492 CAS.
- M. Tan, X. Han, S. Ru, C. Zhang, Z. Ji, Z. Shi, G. Qiao, Y. Wang, R. Cui, Q. Luo, J. Jiao, Y. Li and T. Lu, Nano Res., 2023, 16, 2059–2064 CrossRef CAS.
- T.-H. Wang, C.-Y. Lin, Y.-C. Huang and C.-Y. Li, Electrochim. Acta, 2023, 437, 141500 CrossRef CAS.
- Y. Shi, M. Hou, J. Li, L. Li and Z. Zhang, Acta Phys.-Chim. Sin., 2022, 38, 2206020 Search PubMed.
- M. Hou, Y. X. Shi, J. J. Li, Z. Gao and Z. Zhang, Chem. – Asian J., 2022, 17, e202200624 CAS.
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mat., 2020, 19, 266–276 CrossRef CAS PubMed.
- J. H. Lee, S. Kattel, Z. Xie, B. M. Tackett, J. Wang, C.-J. Liu and J. G. Chen, Adv. Funct. Mater., 2018, 28, 1804762 CrossRef.
- K. Albrecht, K. Matsuoka, K. Fujita and K. Yamamoto, Angew. Chem., Int. Ed., 2015, 54, 5677–5682 CrossRef CAS PubMed.
- K. Albrecht and K. Yamamoto, J. Am. Chem. Soc., 2009, 131, 2244–2251 CrossRef CAS PubMed.
- N. D. McClenaghan, R. Passalacqua, F. Loiseau, S. Campagna, B. Verheyde, A. Hameurlaine and W. Dehaen, J. Am. Chem. Soc., 2003, 125, 5356–5365 CrossRef PubMed.
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef CAS PubMed.
- N. Mayet, K. Servat, K. B. Kokoh and T. W. Napporn, Surfaces, 2019, 2, 257–276 CrossRef CAS.
- Z. Cao, S. B. Zacate, X. Sun, J. Liu, E. M. Hale, W. P. Carson, S. B. Tyndall, J. Xu, X. Liu, X. Liu, C. Song, J.-h Luo, M.-J. Cheng, X. Wen and W. Liu, Angew. Chem., Int. Ed., 2018, 57, 12675–12679 CrossRef CAS PubMed.
- M. Ayiania, M. Smith, A. J. R. Hensley, L. Scudiero, J.-S. McEwen and M. Garcia-Perez, Carbon, 2020, 162, 528–544 CrossRef CAS.
- P. Ledwon, Org. Electron., 2019, 75, 105422 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc00350g |
| This journal is © The Royal Society of Chemistry 2023 |