
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral carbonyl hypoiodites†
Milla
Mattila
 ,
Kari
Rissanen
* and
Jas S.
Ward
*
,
Kari
Rissanen
* and
Jas S.
Ward
*
University of Jyvaskyla, Department of Chemistry, Jyväskylä 40014, Finland. E-mail: kari.t.rissanen@jyu.fi; james.s.ward@jyu.fi
First published on 24th March 2023
Abstract
Three chiral carbonyl hypoiodites, R–C(O)OI, have been prepared from N-protected (S)-valine to give the ligand-stabilised (S)-valinoyl hypoiodite complexes with 4-dimethylaminopyridine, 4-pyrrolidinopyridine, and 4-morpholinopyridine as the stabilising ligands. The identity of the complexes was established by NMR (1H, 13C, 1H–15N HMBC) and single crystal X-ray diffraction analysis.
Weak intermolecular interactions comprise all chemical entities, though halogen bonding (XB) has only recently been defined as the attraction between an electrophilic halogen atom and a neutral or anionic nucleophile,1 and systematically studied as a key interaction. The supramolecular research has previously focused on the construction of non-covalent systems using hydrogen bonding or through metal coordination, but over the last two decades halogen bonding has expanded rapidly as a field of research,2–4 and now is one of the most studied non-covalent interactions.
Halogen(I) (or halenium) ions are the extreme case of a fully ionised halogen atom to a formally positive ion that is stabilised by a pair of Lewis bases (L; usually nitrogen-based) in the form [L–X–L]+ (X = Cl, Br, I; also termed a halonium complex),5–8 and as such fall under the umbrella of complexes that exhibit strong halogen bonds, even though their reactivity differs greatly from other classical XB complexes.4 Due to the origin of XB as a σ-hole interaction,4 which bestows a reliable high degree of linear directionality in its behaviour, halogen(I) ions have found great utility in self-assembling supramolecular architectures.9–11
Prof. Jose Barluenga's eponymous reagent, [bis(pyridine)iodine(I)]BF4 (Barluenga's Reagent, BR) has enjoyed widespread use as a mild iodinating species and oxidant since the 1990s, making it the quintessential iodine(I) reagent.12–14 Notable uses of BR include the iodination of alkenes, alkynes, and arenes under mild conditions, the conversion of 1,2-diols to their respective dicarbonyls, and due to being mild enough, the selective iodination of tyrosine in peptides or proteins, as well as the activation of thio- and n-pentenyl glycosides for glycosylation or conversion to the corresponding glycosyl fluorides.15 However, these studies were performed with an unrestrained homoleptic iodine(I) complex (BR), with the chiral iodine(I) complexes only being studied only as their restrained, bidentate complexes,16 which is also hindered by their availability (or lack thereof).17
Whilst heteroleptic iodine(I) complexes have been confirmed in the solid state,18 the observation of them undergoing ligand scrambling in solution has stifled potential studies into their use.18,19 These results negate synthetic strategies toward creating heteroleptic iodine(I) complexes via the use of chiral ligands, given that the unwanted recovery of meso isomers is highly probable. However, strategies involving the use of bidentate ligands have successfully circumvented these issues,7,16,20 but often entails extensive synthetic protocols, and the influence of the chelation effect of these bidentate ligands on the reactivity cannot be overlooked.
The recent revitalisation of carbonyl hypoiodites,17,21–23 as seen from the viewpoint of halogen bonding as isolable iodine(I) complexes, offers an alternative to the traditional [N–I–N]+ complexes like BR. The carbonyl hypoiodites are inherently heteroleptic, but can still be synthesised in an analogous fashion via a similar Ag+ to I+ cation exchange as the [N–I–N]+ complexes. Until now, alkyl hypoiodites (R–OI; R = alkyl) and carbonyl hypoiodites (R–C(O)OI, R = alkyl, aryl) have almost entirely been used as in situ reagents.24 These hypoiodites have been reported to perform a variety of exotic transformations,25,26 as well as recently being confirmed to perform the same straightforward iodinations that BR can.17 Discerning structure–reactivity relationships can be difficult though, as such hypoiodite reagents like CH3C(O)OI and tBuOI have only limited solution-state data available in the literature,27,28 with the identities of the actual reactive species being unresolved.29,30
Herein, the first examples of chiral carbonyl hypoiodite complexes from N-protected (S)-valine are reported. The availability of α-amino acids as inexpensive, enantiopure starting materials is ubiquitous in organic synthesis, making them ideal chemical feedstocks. However, the double functionality of amino acids makes them incompatible with the Ag+ to I+ cation exchange necessary to produce the desired C(O)O–I–N carbonyl hypoiodites, therefore, (S)-valine was converted to (S)-N-phthaloylvaline (1; Scheme 1) via a double condensation reaction using phthalic anhydride under melt conditions as per a literature protocol.31 Subsequent to the N-phthaloyl protection, 1 could be treated as a straightforward carboxylic acid (analogous to previous carbonyl hypoiodite studies21–23) and was converted to Ag[(S)-N-phthaloylvalinate] (2; Scheme 1) via deprotonation with NaOH, followed by cation exchange with AgNO3 in 43% yield (Scheme 1). The reaction of 2 in dichloromethane with elemental iodine and a pyridine-based ligand, either 4-dimethylaminopyridine (DMAP), 4-pyrrolidinopyridine (4-pyrpy), or 4-morpholinopyridine (4-morpy), gave the carbonyl hypoiodites DMAP (S)-N-phthaloylvalinoyl hypoiodite (3a), 4-pyrpy (S)-N-phthaloylvalinoyl hypoiodite (3b), 4-morpy (S)-N-phthaloylvalinoyl hypoiodite (3c) (Scheme 1). All the hypoiodite complexes (3a–3c) could be isolated as pure solids in 65% to 91% yields, ideal for prospective reagents,17 which can be a weakness of highly reactive iodine(I) complexes.8,32 The synthesis of the analogous derivative with pyridine, (S)-N-phthaloylvalinoyl hypoiodite(pyridine), was attempted given that it would be the directly comparable to BR. However, NMR studies immediately after synthesis of the compound revealed decomposition from its inception.
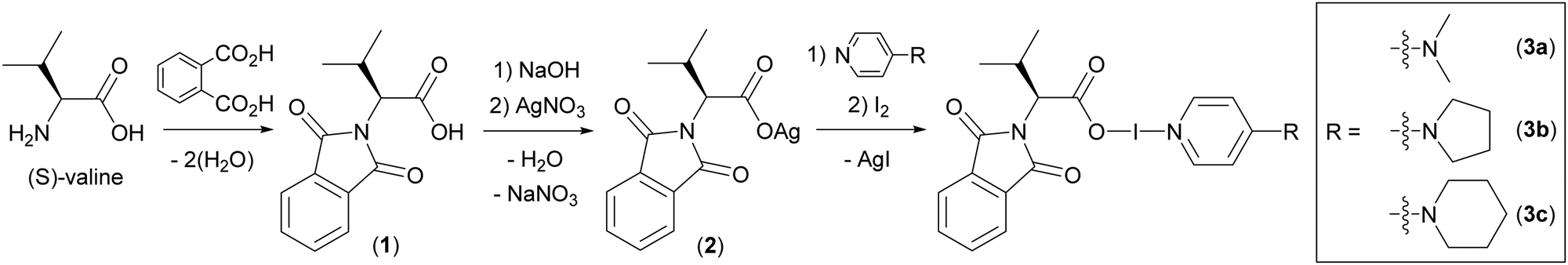 | ||
| Scheme 1 The reaction protocols used to synthesis the hypoiodite compounds (3a–3c) in three steps starting from commercially available (S)-valine. | ||
The complexes 1–3(a–c) were studied in solution by 1H, 13C, and 1H-15N HMBC NMR spectroscopy. However, the poor solubility of 2 necessitated that strongly polar (CD3)2SO be used, unlike 1 and 3a–3c which favoured organic solvents, making direct comparisons of 2 with the hypoiodite compounds (3a–3c) ambiguous. Nevertheless, the comparison of the free ligand 4-morpy,23 N-protected amino acid 1, and 3c (Fig. 1) did reveal the general trend of the 3c1H NMR signals moving upfield. The pyridinic hydrogen atoms of 3c (Ha and Hb; compared to 4-morpy) and the hydrogen atoms of the N-protected valine moiety (He, Hf, Hg, Hh, Hi, Hj; compared to 1) were both shifted upfield with noticeable, but modest, differences all within the range of 0.02–0.13 ppm (for 3c compared to 4-morpy and 1). The same general trends were also observed for 3a (with free DMAP and 1) and 3b (with free 4-pyrpy and 1), with 3a demonstrating the largest coordination shift of 0.23 ppm for its respective α-hydrogen atom (cf. He in Fig. 1) in comparison to 1. Interestingly, this is contrary to the general trend observed for [N–I–N]+ complexes where the I+ complexes experience a downfield shift of their 1H NMR peaks compared to their respective free ligands and linear [N–Ag–N]+ precursor complexes.8,33
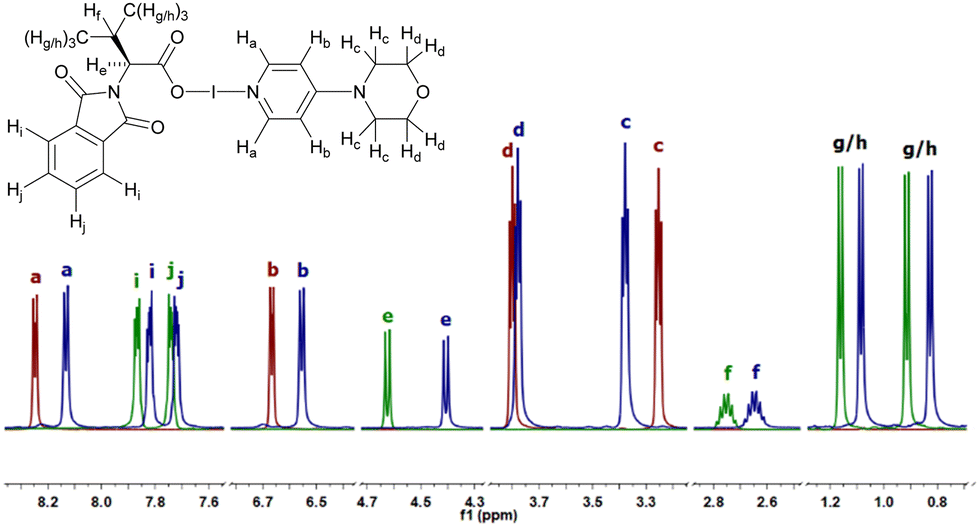 | ||
| Fig. 1 The superimposed and condensed 1H NMR spectra of the free ligand 4-morpy (red), the N-protected valine 1 (green), and 4-morpy hypoiodite 3c (blue), showing the general trend of the hypoiodite shifting upfield compared to its constituent components (inset: Annotated molecule of 3c; hydrogen labels for the subcomponents, 4-morpy and 1, follow the same nomenclature as 3c). | ||
The 1H–15N HMBC studies in CD2Cl2 were used to determine the 15N NMR chemical shifts of the pyridinic nitrogen atoms, with those being aptly positioned to reflect the characteristic iodine(I) to nitrogen interaction upon complexation. The 15N NMR chemical shifts of the pyridinic nitrogen atoms of −212.5 ppm (3a), −214.7 ppm (3b), and −204.2 ppm (3c), all demonstrated significant, and characteristically indicative, coordination shifts (ΔδN; defined as δ[15Nhypoiodite]-δ[15NL]) from their respective free ligands (L = DMAP: −108.6 ppm;21 4-pyrpy: −110.0 ppm; 4-morpy: −99.1 ppm23) of −103.9 ppm (DMAP to 3a), −104.7 ppm (4-pyrpy to 3b), and −105.1 ppm (4-morpy to 3c) upon formation of the carbonyl hypoiodite complexes. These ΔδN values were in line with previous observations for other carbonyl hypoiodite complexes,21–23 as were the values of the 15N NMR chemical shifts themselves which fell within the range of −150 to −250 ppm for reported O–I–N carbonyl hypoiodite compounds (cf. −209.5 ppm for PhC(O)OI-(DMAP) and −202.1 ppm for PhC(O)OI-(4-morpy); both in CD2Cl2).21 Whilst the phthalimido 15N NMR chemical shifts were observed in high concentration samples of 1 (−219.4 ppm) and 2 (−212.9 ppm), the phthalimido 15N NMR peaks were not observed for 3a–3c, which not surprising given that the HMBC parameters were optimised for the more informative pyridine-based ligands, and with the possibility of the much weaker phthalimido resonances being obscured under the stronger pair of ligand resonances present in 3a–3c.
The identity of the hypoiodite compounds 3a–3c were also established by single crystal X-ray diffraction (SCXRD; Fig. 2), which confirmed their chirality with the non-centrosymmetric space groups P21 (3a), P1 (3b), and I2 (3c). Interestingly, all three compounds were found to have two independent molecules in the asymmetric unit cell (all with S-stereochemistry), with the slight differences observed in the bonding parameters between each pair being ultimately within the error of the measurements to a 3σ tolerance.‡ The I–N and I–O bond lengths of 2.26(2)/2.21(2) and 2.21(1)/2.24(1) Å (3a), 2.243(6)/2.235(7) and 2.209(6)/2.237(6) Å (3b), 2.24(1)/2.25(1) and 2.22(1)/2.27(1) Å (3c), respectively, are comparable to those of similar carbonyl hypoiodite complexes previously reported based on simple carboxylic acids (cf. MeC(O)OI-(DMAP): I–N = 2.24(1) Å and I–O = 2.20(1) Å and PhC(O)OI-(DMAP): I–N = 2.241(3) Å and I–O = 2.210(3) Å). The O–I–N bond angles of 172.6(5)/175.2(5)° (3a), 177.8(2)/173.7(2)° (3b), and 171.8(4)/175.7(4)° (3c), reflect the strong preference toward linearity enforced by the halogen bonding, though are some of the largest known deviations from 180° observed for carbonyl hypoiodites (cf. MeC(O)OI-(DMAP): O–I–N = 172.9(5)°; PhC(O)OI-(DMAP): O–I–N = 178.1(1)°), possibly due to 3a–3c possessing the most sterically encumbered substituents of known carbonyl hypoiodite compounds with the presence of the phthaloyl and iPr groups at the α-position of the N-protected amino acid backbone. The packing of 3a–3c revealed no intermolecular or close-proximity interactions with the I+⋯I+ distances for all compounds being greater than 4.81 Å (cf. combined van der Waals radii for I⋯I = 3.96 Å).
 | ||
| Fig. 2 The molecular structures of 3a (top), 3b (middle), and 3c (bottom), annotated with their O–I–N angles (°) and I–N/I–O bond lengths (Å) (minor disordered positions of 3a and 3b omitted for clarity; thermal parameters at 50% probability; only one of each pair of crystallographically independent molecules shown for 3a–3c, with parameters for the omitted second molecule in each case annotated in square brackets). | ||
In conclusion, the first chiral carbonyl hypoiodite complexes have been synthesised, based on the cheap and abundantly available enantiopure amino acid (S)-valine. In only a three-step reaction, (S)-valine was N-protected to 1, its carboxylic acid transformed into the prerequisite silver(I) carboxylate precursor 2, and finally converted into the desired carbonyl hypoiodite complexes with a variety of pyridine-based stabilising ligands (3a–3c). The complexes were found to demonstrate relatively good stability for iodine(I) species, permitting full NMR and SCXRD studies, and enabling them to be isolated as pure solids. Despite their sterically more encumbered substituents, the chiral carbonyl hypoiodite complexes 3a–3c displayed NMR and crystallographic metrics comparable to other previously reported carbonyl hypoiodites. The carbonyl hypoiodites 3a–3c represent an elegant workaround to the potential complications of chiral [N–I–N]+ complexes analogous to Barluenga's reagent (which suffer from ligand scrambling), and opens up a new avenue of approach toward potentially enantioselective iodination reactions, with studies currently ongoing to this end.
The authors gratefully acknowledge the Academy of Finland (K.R. grant No. 351121), the Magnus Ehrnrooth Foundation (J.S.W.), and the University of Jyväskylä, Finland for financial support.
M.M. and J.S.W. were responsible for the preparation of the compounds and crystallographic samples, as well as conducting the solution studies. J.S.W. and K.R. was responsible for the conceptualisation of the research and provided supervision, with J.S.W. performing the X-ray studies. The visualisation and writing of the manuscript were undertaken by J.S.W., with K.R. and J.S.W. performing the reviewing and editing. K.R. and J.S.W. were responsible for the acquisition of funding.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- M. H. Kolář and P. Hobza, Chem. Rev., 2016, 116, 5155–5187 CrossRef PubMed.
- J. Pancholi and P. D. Beer, Coord. Chem. Rev., 2020, 416, 213281 CrossRef CAS.
- D. von der Heiden, A. Vanderkooy and M. Erdélyi, Coord. Chem. Rev., 2020, 407, 213147 CrossRef CAS.
- J. S. Ward, K.-N. Truong, M. Erdélyi and K. Rissanen, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2022 DOI:10.1016/B978-0-12-823144-9.00043-1.
- A.-C. C. Carlsson, K. Mehmeti, M. Uhrbom, A. Karim, M. Bedin, R. Puttreddy, R. Kleinmaier, A. A. Neverov, B. Nekoueishahraki, J. Gräfenstein, K. Rissanen and M. Erdélyi, J. Am. Chem. Soc., 2016, 138, 9853–9863 CrossRef CAS PubMed.
- J. S. Ward, A. Frontera and K. Rissanen, Dalton Trans., 2021, 50, 8297–8301 RSC.
- L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, Angew. Chem., Int. Ed., 2016, 55, 14033–14036 CrossRef CAS PubMed.
- L. Turunen, U. Warzok, C. A. Schalley and K. Rissanen, Chemistry, 2017, 3, 861–869 CrossRef CAS.
- A. Vanderkooy, A. K. Gupta, T. Földes, S. Lindblad, A. Orthaber, I. Pápai and M. Erdélyi, Angew. Chem., Int. Ed., 2019, 58, 9012–9016 CrossRef CAS PubMed.
- J. Barluenga, J. M. González, M. A. Garcia-Martin, P. J. Campos and G. Asensio, J. Chem. Soc., Chem. Commun., 1992, 1016–1017 RSC.
- J. Ezquerra, C. Pedregal, C. Lamas, J. Barluenga, M. Pérez, M. A. García-Martín and J. M. González, J. Org. Chem., 1996, 61, 5804–5812 CrossRef CAS.
- G. Espuña, G. Arsequell, G. Valencia, J. Barluenga, M. Pérez and J. M. González, Chem. Commun., 2000, 1307–1308 RSC.
- J. M. Chalker, A. L. Thompson, B. G. Davis, N. Zaware and P. Wipf, Org. Synth., 2010, 87, 288–298 CrossRef CAS.
- D. von der Heiden, F. B. Németh, M. Andreasson, D. Sethio, I. Pápai and M. Erdélyi, Org. Biomol. Chem., 2021, 19, 8307–8323 RSC.
- L. M. E. Wilson, K. Rissanen and J. S. Ward, New J. Chem., 2023, 47, 2978–2982 RSC.
- J. S. Ward, G. Fiorini, A. Frontera and K. Rissanen, Chem. Commun., 2020, 56, 8428–8431 RSC.
- D. von der Heiden, K. Rissanen and M. Erdélyi, Chem. Commun., 2020, 56, 14431–14434 RSC.
- S. Lindblad, F. B. Németh, T. Földes, D. von der Heiden, H. G. Vang, Z. L. Driscoll, E. R. Gonnering, I. Pápai, N. Bowling and M. Erdélyi, Chem. – Eur. J., 2021, 27, 13748–13756 CrossRef CAS PubMed.
- S. Yu, J. S. Ward, K.-N. Truong and K. Rissanen, Angew. Chem., Int. Ed., 2021, 60, 20739–20743 CrossRef CAS PubMed.
- E. Kramer, S. Yu, J. S. Ward and K. Rissanen, Dalton Trans., 2021, 50, 14990–14993 RSC.
- J. S. Ward, J. Martõnova, L. M. E. Wilson, E. Kramer, R. Aav and K. Rissanen, Dalton Trans., 2022, 51, 14646–14653 RSC.
- R. Kawasumi, S. Narita, K. Miyamoto, K. Tominaga, R. Takita and M. Uchiyama, Sci. Rep., 2017, 7, 17967 CrossRef PubMed.
- D. D. Tanner and G. C. Gidley, J. Am. Chem. Soc., 1968, 90, 808–809 CrossRef CAS.
- J. Barluenga, F. González-Bobes and J. M. González, Angew. Chem., Int. Ed., 2002, 41, 2556–2558 CrossRef CAS PubMed.
- T. Hokamp, A. T. Storm, M. Yusubov and T. Wirth, Synlett, 2018, 415–418 CAS.
- J. L. Courtneidge, J. Lusztyk and D. Pagé, Tetrahedron Lett., 1994, 35, 1003–1006 CrossRef CAS.
- R. Montoro and T. Wirth, Org. Lett., 2003, 5, 4729–4731 CrossRef CAS PubMed.
- D. D. Tanner, G. C. Gidley, N. Das, J. E. Rowe and A. Potter, J. Am. Chem. Soc., 1984, 106, 5261–5267 CrossRef CAS.
- A. G. Griesbeck, H. Mauder and I. Müller, Chem. Ber., 1992, 125, 2467–2475 CrossRef CAS.
- J. S. Ward, R. M. Gomila, A. Frontera and K. Rissanen, RSC Adv., 2022, 12, 8674–8682 RSC.
- J. S. Ward, CrystEngComm, 2022, 24, 7029–7033 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, NMR, and X-ray. CCDC 2225069–2225071, 2245212. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc00259d |
| ‡ With the exception of the I–O bond lengths (2.22(1)/2.27(1) Å) between the pair of independent molecules in 3c, which are just beyond a 3σ tolerance by less than 0.01 Å. |
| This journal is © The Royal Society of Chemistry 2023 |